Applied Bioinformatics Laboratory, the University of Kansas, Lawrence, KS 66047, USA.
BMC Bioinformatics. 2010 Jan 28;11:62. doi: 10.1186/1471-2105-11-62.
The ability to design thermostable proteins is theoretically important and practically useful. Robust and accurate algorithms, however, remain elusive. One critical problem is the lack of reliable methods to estimate the relative thermostability of possible mutants.
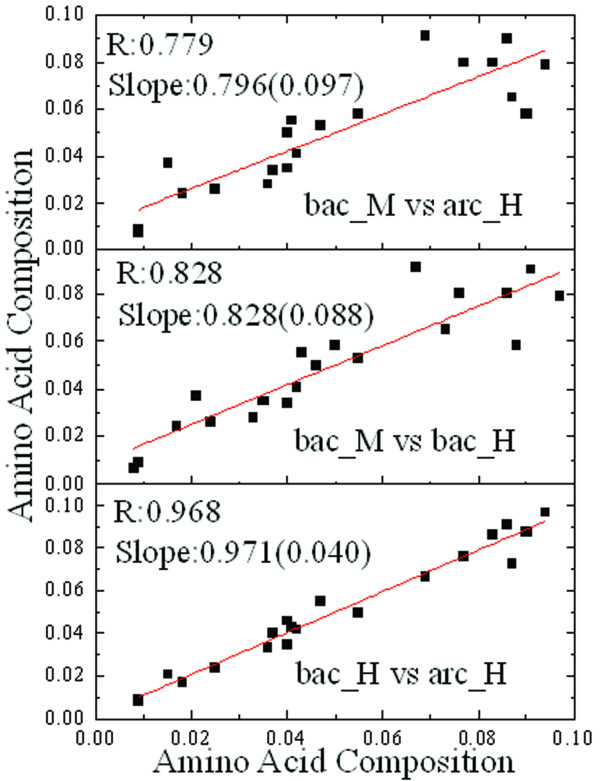
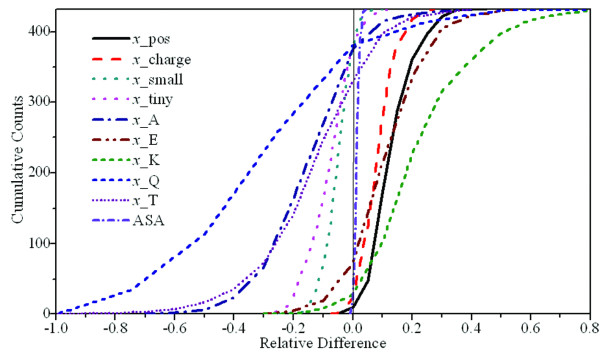
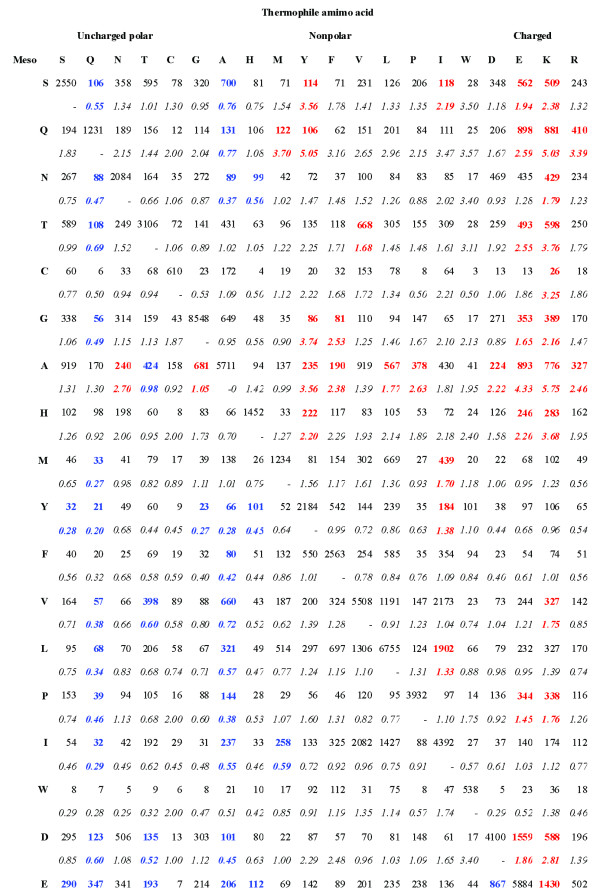
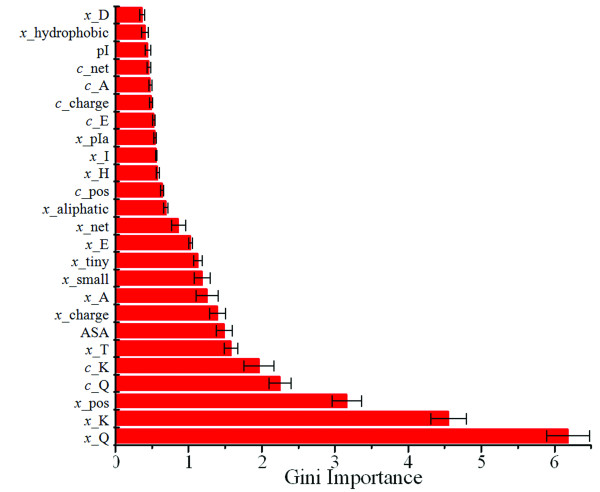
We report a novel scoring function for discriminating hyperthermophilic and mesophilic proteins with application to predicting the relative thermostability of protein mutants. The scoring function was developed based on an elaborate analysis of a set of features calculated or predicted from 540 pairs of hyperthermophilic and mesophilic protein ortholog sequences. It was constructed by a linear combination of ten important features identified by a feature ranking procedure based on the random forest classification algorithm. The weights of these features in the scoring function were fitted by a hill-climbing algorithm. This scoring function has shown an excellent ability to discriminate hyperthermophilic from mesophilic sequences. The prediction accuracies reached 98.9% and 97.3% in discriminating orthologous pairs in training and the holdout testing datasets, respectively. Moreover, the scoring function can distinguish non-homologous sequences with an accuracy of 88.4%. Additional blind tests using two datasets of experimentally investigated mutations demonstrated that the scoring function can be used to predict the relative thermostability of proteins and their mutants at very high accuracies (92.9% and 94.4%). We also developed an amino acid substitution preference matrix between mesophilic and hyperthermophilic proteins, which may be useful in designing more thermostable proteins.
We have presented a novel scoring function which can distinguish not only HP/MP ortholog pairs, but also non-homologous pairs at high accuracies. Most importantly, it can be used to accurately predict the relative stability of proteins and their mutants, as demonstrated in two blind tests. In addition, the residue substitution preference matrix assembled in this study may reflect the thermal adaptation induced substitution biases. A web server implementing the scoring function and the dataset used in this study are freely available at http://www.abl.ku.edu/thermorank/.
设计热稳定蛋白在理论上很重要,在实践中也很有用。然而,稳健且准确的算法仍然难以实现。一个关键问题是缺乏可靠的方法来估计可能突变体的相对热稳定性。
我们报告了一种用于区分高热和中温蛋白的新型评分函数,并将其应用于预测蛋白质突变体的相对热稳定性。该评分函数是基于对 540 对高热和中温蛋白直系同源序列计算或预测的一组特征的精心分析而开发的。它是通过基于随机森林分类算法的特征排序过程确定的十个重要特征的线性组合构建的。在评分函数中的这些特征的权重通过爬山算法拟合。该评分函数在区分高热和中温序列方面表现出出色的能力。在训练和保留测试数据集的直系同源对中,预测准确率分别达到 98.9%和 97.3%。此外,该评分函数可以以 88.4%的准确率区分非同源序列。使用两个经过实验研究的突变数据集进行的额外盲测表明,该评分函数可用于以非常高的准确度预测蛋白质及其突变体的相对热稳定性(92.9%和 94.4%)。我们还开发了一种在中温和高热蛋白之间的氨基酸取代偏好矩阵,这可能有助于设计更耐热的蛋白质。
我们提出了一种新的评分函数,不仅可以区分 HP/MP 直系同源对,而且可以以高精度区分非同源对。最重要的是,如两个盲测所示,它可用于准确预测蛋白质及其突变体的相对稳定性。此外,本研究中组装的残基取代偏好矩阵可能反映了热适应诱导的取代偏差。实现评分函数和本研究中使用的数据集的网络服务器可在 http://www.abl.ku.edu/thermorank/ 上免费获得。