Department of Computer Science, National University of Singapore, Singapore.
BMC Bioinformatics. 2010 Jan 29;11:66. doi: 10.1186/1471-2105-11-66.
Human genome contains millions of common single nucleotide polymorphisms (SNPs) and these SNPs play an important role in understanding the association between genetic variations and human diseases. Many SNPs show correlated genotypes, or linkage disequilibrium (LD), thus it is not necessary to genotype all SNPs for association study. Many algorithms have been developed to find a small subset of SNPs called tag SNPs that are sufficient to infer all the other SNPs. Algorithms based on the r2 LD statistic have gained popularity because r2 is directly related to statistical power to detect disease associations. Most of existing r2 based algorithms use pairwise LD. Recent studies show that multi-marker LD can help further reduce the number of tag SNPs. However, existing tag SNP selection algorithms based on multi-marker LD are both time-consuming and memory-consuming. They cannot work on chromosomes containing more than 100 k SNPs using length-3 tagging rules.
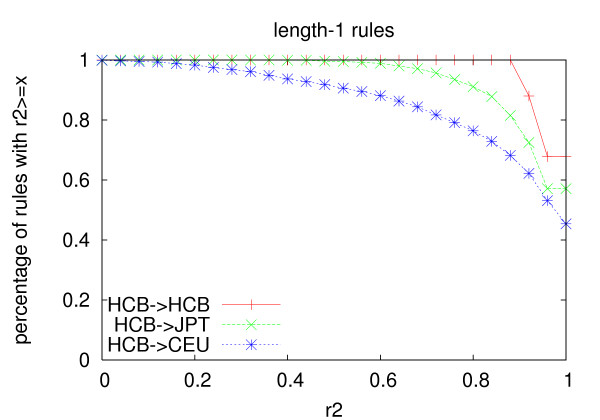
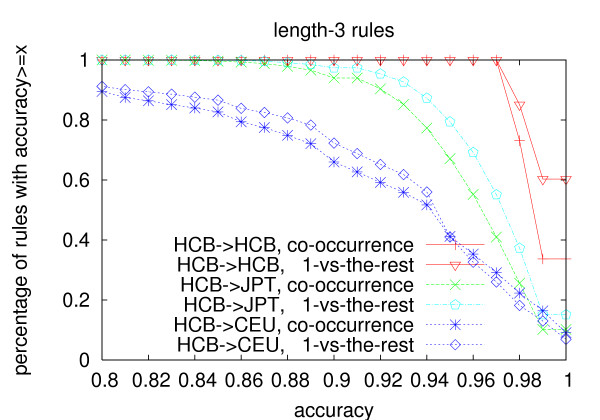
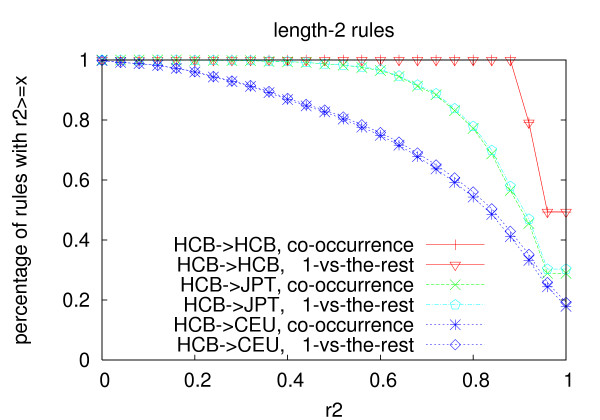
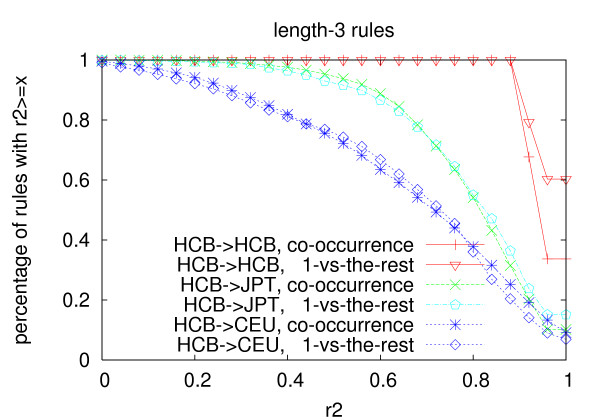
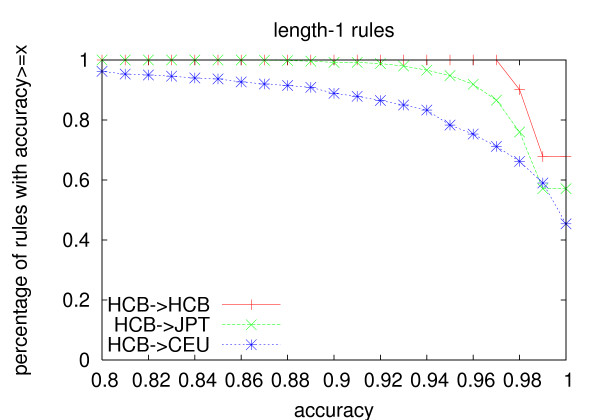
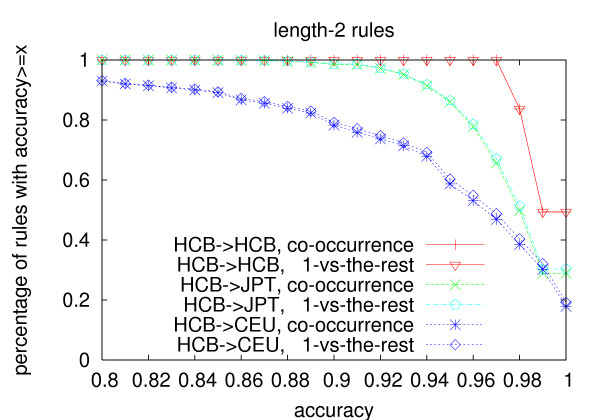
We propose an efficient algorithm called FastTagger to calculate multi-marker tagging rules and select tag SNPs based on multi-marker LD. FastTagger uses several techniques to reduce running time and memory consumption. Our experiment results show that FastTagger is several times faster than existing multi-marker based tag SNP selection algorithms, and it consumes much less memory at the same time. As a result, FastTagger can work on chromosomes containing more than 100 k SNPs using length-3 tagging rules.FastTagger also produces smaller sets of tag SNPs than existing multi-marker based algorithms, and the reduction ratio ranges from 3%-9% when length-3 tagging rules are used. The generated tagging rules can also be used for genotype imputation. We studied the prediction accuracy of individual rules, and the average accuracy is above 96% when r2 >/= 0.9.
Generating multi-marker tagging rules is a computation intensive task, and it is the bottleneck of existing multi-marker based tag SNP selection methods. FastTagger is a practical and scalable algorithm to solve this problem.
人类基因组包含数百万个常见的单核苷酸多态性(SNP),这些 SNP 在理解遗传变异与人类疾病之间的关联方面发挥着重要作用。许多 SNP 显示出相关的基因型,或连锁不平衡(LD),因此没有必要对所有 SNP 进行基因型分析以进行关联研究。已经开发了许多算法来找到一小部分被称为标签 SNP 的 SNP,这些 SNP 足以推断所有其他 SNP。基于 r2 LD 统计量的算法因其与检测疾病关联的统计能力直接相关而受到欢迎。大多数现有的基于 r2 的算法使用成对 LD。最近的研究表明,多标记 LD 可以帮助进一步减少标签 SNP 的数量。然而,现有的基于多标记 LD 的标签 SNP 选择算法既耗时又耗内存。它们不能在使用长度为 3 的标记规则的包含超过 100 k SNP 的染色体上运行。
我们提出了一种名为 FastTagger 的高效算法,用于计算多标记标记规则并基于多标记 LD 选择标签 SNP。FastTagger 使用了几种技术来减少运行时间和内存消耗。我们的实验结果表明,FastTagger 比现有的基于多标记的标签 SNP 选择算法快几倍,同时消耗的内存也少得多。因此,FastTagger 可以在使用长度为 3 的标记规则的包含超过 100 k SNP 的染色体上运行。FastTagger 还生成了比现有的基于多标记的算法更少的标签 SNP 集,当使用长度为 3 的标记规则时,减少比例范围为 3%-9%。生成的标记规则也可用于基因型推断。我们研究了单个规则的预测准确性,当 r2≥0.9 时,平均准确性高于 96%。
生成多标记标记规则是一项计算密集型任务,是现有基于多标记的标签 SNP 选择方法的瓶颈。FastTagger 是解决此问题的实用且可扩展的算法。