Program in Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles CA 90089, USA.
BMC Bioinformatics. 2010 Jan 18;11 Suppl 1(Suppl 1):S62. doi: 10.1186/1471-2105-11-S1-S62.
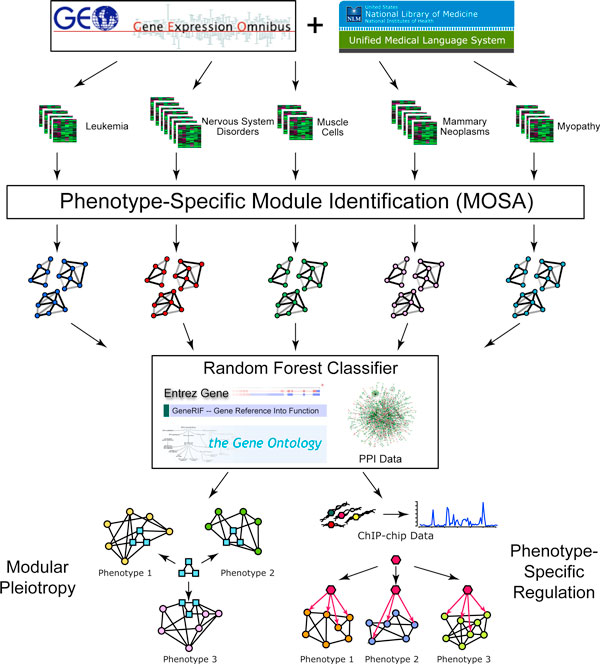
Complex human diseases are often caused by multiple mutations, each of which contributes only a minor effect to the disease phenotype. To study the basis for these complex phenotypes, we developed a network-based approach to identify coexpression modules specifically activated in particular phenotypes. We integrated these modules, protein-protein interaction data, Gene Ontology annotations, and our database of gene-phenotype associations derived from literature to predict novel human gene-phenotype associations. Our systematic predictions provide us with the opportunity to perform a global analysis of human gene pleiotropy and its underlying regulatory mechanisms.
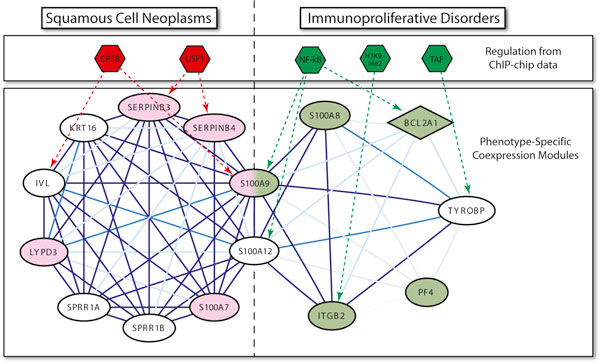
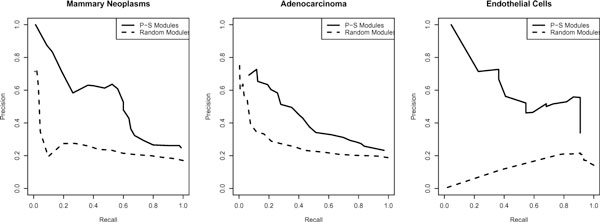
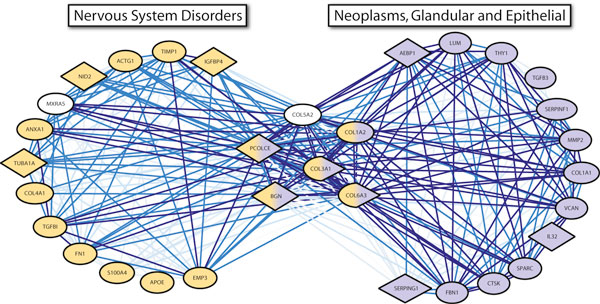
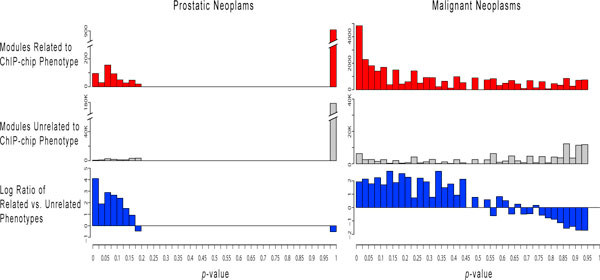
We applied this method to 338 microarray datasets, covering 178 phenotype classes, and identified 193,145 phenotype-specific coexpression modules. We trained random forest classifiers for each phenotype and predicted a total of 6,558 gene-phenotype associations. We showed that 40.9% genes are pleiotropic, highlighting that pleiotropy is more prevalent than previously expected. We collected 77 ChIP-chip datasets studying 69 transcription factors binding over 16,000 targets under various phenotypic conditions. Utilizing this unique data source, we confirmed that dynamic transcriptional regulation is an important force driving the formation of phenotype specific gene modules.
We created a genome-wide gene to phenotype mapping that has many potential implications, including providing potential new drug targets and uncovering the basis for human disease phenotypes. Our analysis of these phenotype-specific coexpression modules reveals a high prevalence of gene pleiotropy, and suggests that phenotype-specific transcription factor binding may contribute to phenotypic diversity. All resources from our study are made freely available on our online Phenotype Prediction Database.
复杂的人类疾病通常是由多个突变引起的,每个突变对疾病表型的贡献都很小。为了研究这些复杂表型的基础,我们开发了一种基于网络的方法来识别特定表型中特异性激活的共表达模块。我们整合了这些模块、蛋白质-蛋白质相互作用数据、GO 注释以及我们从文献中提取的基因-表型关联数据库,以预测新的人类基因-表型关联。我们的系统预测为我们提供了分析人类基因多效性及其潜在调控机制的机会。
我们将这种方法应用于 338 个微阵列数据集,涵盖 178 种表型类别,鉴定出 193145 个表型特异性共表达模块。我们为每个表型训练了随机森林分类器,并预测了总共 6558 个基因-表型关联。我们发现 40.9%的基因是多效性的,这表明多效性比以前预期的更为普遍。我们收集了 77 个 ChIP-chip 数据集,这些数据集研究了 69 个转录因子在各种表型条件下对超过 16000 个靶标的结合情况。利用这个独特的数据源,我们证实了动态转录调控是形成表型特异性基因模块的重要力量。
我们创建了一个全基因组基因到表型的映射,这具有许多潜在的意义,包括提供潜在的新药物靶点,并揭示人类疾病表型的基础。我们对这些表型特异性共表达模块的分析表明基因多效性的发生率很高,并表明表型特异性转录因子结合可能有助于表型多样性。我们研究的所有资源都在我们的在线表型预测数据库中免费提供。