Center for Biomedical Informatics, The Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA.
BMC Bioinformatics. 2010 Feb 4;11:74. doi: 10.1186/1471-2105-11-74.
Recent studies have shown that copy number variations (CNVs) are frequent in higher eukaryotes and associated with a substantial portion of inherited and acquired risk for various human diseases. The increasing availability of high-resolution genome surveillance platforms provides opportunity for rapidly assessing research and clinical samples for CNV content, as well as for determining the potential pathogenicity of identified variants. However, few informatics tools for accurate and efficient CNV detection and assessment currently exist.
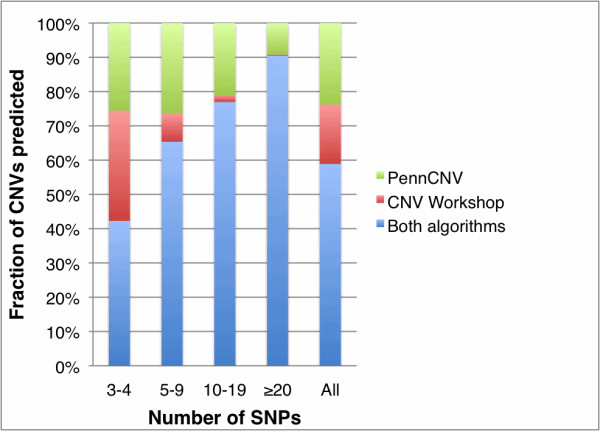
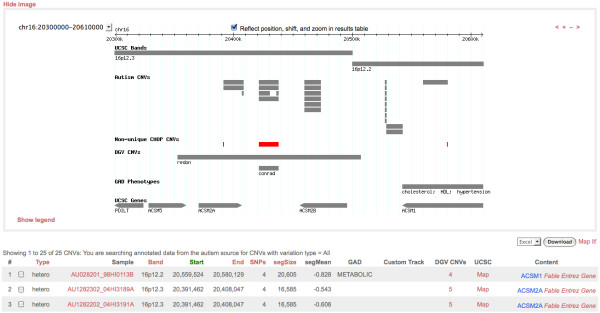
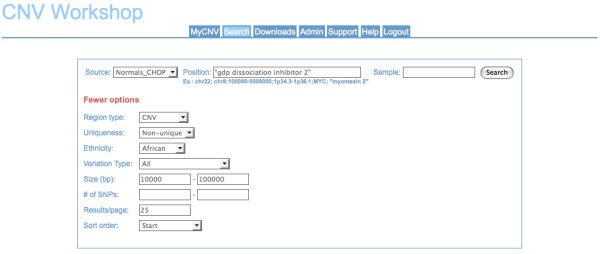
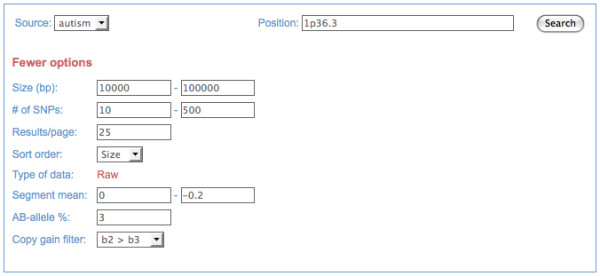
We developed a suite of software tools and resources (CNV Workshop) for automated, genome-wide CNV detection from a variety of SNP array platforms. CNV Workshop includes three major components: detection, annotation, and presentation of structural variants from genome array data. CNV detection utilizes a robust and genotype-specific extension of the Circular Binary Segmentation algorithm, and the use of additional detection algorithms is supported. Predicted CNVs are captured in a MySQL database that supports cohort-based projects and incorporates a secure user authentication layer and user/admin roles. To assist with determination of pathogenicity, detected CNVs are also annotated automatically for gene content, known disease loci, and gene-based literature references. Results are easily queried, sorted, filtered, and visualized via a web-based presentation layer that includes a GBrowse-based graphical representation of CNV content and relevant public data, integration with the UCSC Genome Browser, and tabular displays of genomic attributes for each CNV.
To our knowledge, CNV Workshop represents the first cohesive and convenient platform for detection, annotation, and assessment of the biological and clinical significance of structural variants. CNV Workshop has been successfully utilized for assessment of genomic variation in healthy individuals and disease cohorts and is an ideal platform for coordinating multiple associated projects.
Available on the web at: http://sourceforge.net/projects/cnv.
最近的研究表明,拷贝数变异(CNVs)在高等真核生物中很常见,与多种人类疾病的遗传和获得性风险的很大一部分有关。越来越多的高分辨率基因组监测平台提供了机会,可以快速评估研究和临床样本的 CNV 含量,以及确定已识别变体的潜在致病性。然而,目前用于准确高效地检测和评估 CNV 的计算工具很少。
我们开发了一套软件工具和资源(CNV Workshop),用于从各种 SNP 阵列平台自动进行全基因组 CNV 检测。CNV Workshop 包括三个主要组件:从基因组阵列数据中检测、注释和呈现结构变体。CNV 检测利用了一种强大的、基于基因型的 Circular Binary Segmentation 算法扩展,并且支持使用其他检测算法。预测的 CNV 被捕获在一个 MySQL 数据库中,该数据库支持基于队列的项目,并整合了安全的用户身份验证层和用户/管理员角色。为了帮助确定致病性,检测到的 CNV 还会自动注释基因内容、已知疾病位点和基于基因的文献参考。结果可以通过基于网络的呈现层轻松查询、排序、过滤和可视化,该呈现层包括基于 GBrowse 的 CNV 内容和相关公共数据的图形表示、与 UCSC 基因组浏览器的集成以及每个 CNV 的基因组属性的表格显示。
据我们所知,CNV Workshop 是第一个用于检测、注释和评估结构变体的生物学和临床意义的综合且方便的平台。CNV Workshop 已成功用于评估健康个体和疾病队列的基因组变异,并且是协调多个相关项目的理想平台。
可在以下网址获得:http://sourceforge.net/projects/cnv。