School of Mathematical Sciences, Peking University, Beijing, China.
National Center of Mathematics and Interdisciplinary Sciences, Academy of Mathematics and Systems Science, Chinese Academy of Sciences, Beijing, China.
PLoS One. 2018 Apr 27;13(4):e0196226. doi: 10.1371/journal.pone.0196226. eCollection 2018.
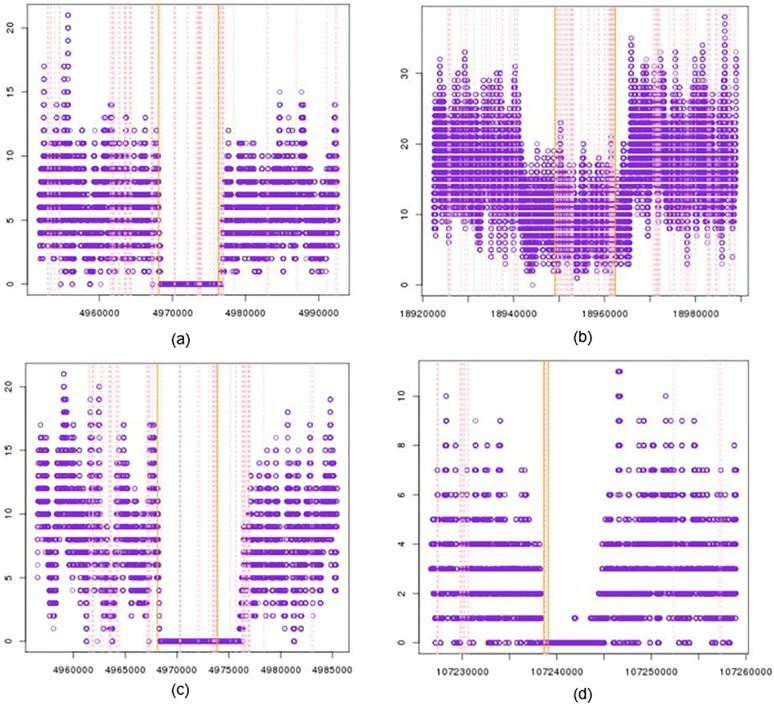
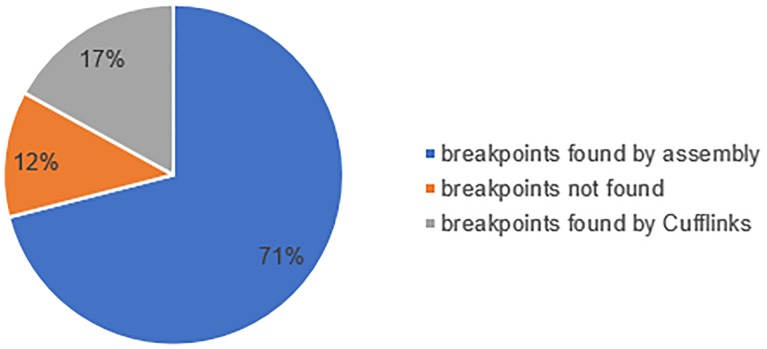
Copy number variations (CNVs) are gain and loss of DNA sequence of a genome. High throughput platforms such as microarrays and next generation sequencing technologies (NGS) have been applied for genome wide copy number losses. Although progress has been made in both approaches, the accuracy and consistency of CNV calling from the two platforms remain in dispute. In this study, we perform a deep analysis on copy number losses on 254 human DNA samples, which have both SNP microarray data and NGS data publicly available from Hapmap Project and 1000 Genomes Project respectively. We show that the copy number losses reported from Hapmap Project and 1000 Genome Project only have < 30% overlap, while these reports are required to have cross-platform (e.g. PCR, microarray and high-throughput sequencing) experimental supporting by their corresponding projects, even though state-of-art calling methods were employed. On the other hand, copy number losses are found directly from HapMap microarray data by an accurate algorithm, i.e. CNVhac, almost all of which have lower read mapping depth in NGS data; furthermore, 88% of which can be supported by the sequences with breakpoint in NGS data. Our results suggest the ability of microarray calling CNVs and the possible introduction of false negatives from the unessential requirement of the additional cross-platform supporting. The inconsistency of CNV reports from Hapmap Project and 1000 Genomes Project might result from the inadequate information containing in microarray data, the inconsistent detection criteria, or the filtration effect of cross-platform supporting. The statistical test on CNVs called from CNVhac show that the microarray data can offer reliable CNV reports, and majority of CNV candidates can be confirmed by raw sequences. Therefore, the CNV candidates given by a good caller could be highly reliable without cross-platform supporting, so additional experimental information should be applied in need instead of necessarily.
拷贝数变异(CNVs)是基因组中 DNA 序列的增益和损失。微阵列和下一代测序技术(NGS)等高通量平台已被用于全基因组拷贝数缺失。尽管这两种方法都取得了进展,但来自这两个平台的 CNV 调用的准确性和一致性仍存在争议。在这项研究中,我们对 254 个人类 DNA 样本的拷贝数缺失进行了深入分析,这些样本分别来自 Hapmap 项目和 1000 基因组项目的 SNP 微阵列数据和 NGS 数据。我们表明,Hapmap 项目和 1000 基因组项目报告的拷贝数缺失<30%,而这些报告需要有交叉平台(如 PCR、微阵列和高通量测序)的实验支持,即使采用了最先进的调用方法。另一方面,通过一种准确的算法,即 CNVhac,直接从 HapMap 微阵列数据中发现拷贝数缺失,几乎所有这些缺失在 NGS 数据中的读取映射深度都较低;此外,其中 88%可以通过 NGS 数据中具有断点的序列来支持。我们的结果表明,微阵列调用 CNVs 的能力以及来自不必要的交叉平台支持的额外要求可能会引入假阴性。Hapmap 项目和 1000 基因组项目的 CNV 报告不一致可能是由于微阵列数据中包含的信息不足、不一致的检测标准或交叉平台支持的过滤效应所致。对 CNVhac 调用的 CNVs 进行的统计检验表明,微阵列数据可以提供可靠的 CNV 报告,并且大多数 CNV 候选者可以通过原始序列得到确认。因此,良好的调用器提供的 CNV 候选者可以高度可靠,而无需交叉平台支持,因此应该根据需要应用额外的实验信息,而不是必须应用。