Department of Microbiology, College of Natural Sciences, Kyungpook National University, Daegu 702-701, Republic of Korea.
Virus Res. 2010 May;149(2):175-82. doi: 10.1016/j.virusres.2010.01.015. Epub 2010 Feb 2.
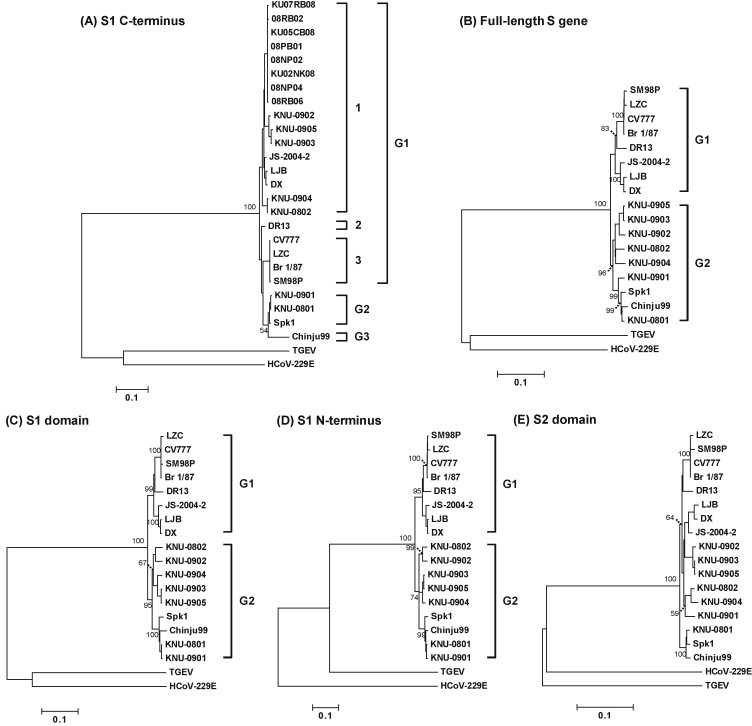
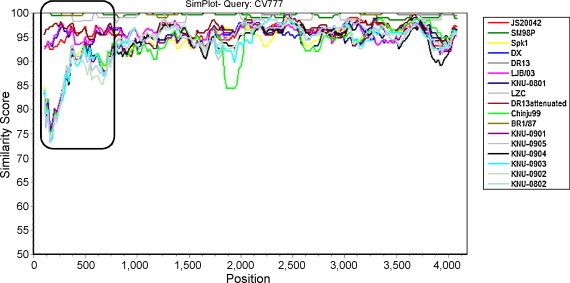
Porcine epidemic diarrhea virus (PEDV) has plagued the domestic swine industry in Korea causing significant economic impacts on pig production nationwide. In the present study, we determined the complete nucleotide sequences of the spike (S) glycoprotein genes of seven Korean PEDV isolates. The entire S genes of all isolates were found to be nine nucleotides longer in length than other PEDV reference strains. This size difference was due to the combined presence of notable 15 bp insertion and 6 bp deletion within the N-terminal region of the S1 domain of the Korean isolates. In addition, the largest number of amino acid variations was accumulated in the S1 N-terminal region, leading to the presence of hypervariability in the isolates. Sequence comparisons at the peptide level of the S proteins revealed that all seven Korean isolates shared diverse similarities ranging from a 93.6% to 99.6% identity with each other but exhibited a 92.2% to 93.7% identity with other reference strains. Collectively, the sequence analysis data indicate the diversity of the PEDV isolates currently prevalent in Korea that represents a heterogeneous group. Phylogenetic analyses showed two separate clusters, in which all Korean field isolates were grouped together in the second cluster (group 2). The results indicate that prevailing isolates in Korea are phylogenetically more closely related to each other rather than other reference strains. Interestingly, the tree topology based on the nucleotide sequences representing the S1 domain or the S1 N-terminal region most nearly resembled the full S gene-based phylogenetic tree. Therefore, our data implicates a potential usefulness of the partial S protein gene including the N-terminal region in unveiling genetic relatedness of PEDV isolates.
猪流行性腹泻病毒(PEDV)一直困扰着韩国的养猪业,给全国的养猪生产带来了重大的经济影响。在本研究中,我们测定了 7 株韩国 PEDV 分离株的 Spike(S)糖蛋白基因的完整核苷酸序列。所有分离株的全长 S 基因比其他 PEDV 参考株长 9 个核苷酸。这种大小差异是由于韩国分离株 S1 结构域的 N 端区域存在显著的 15 个碱基插入和 6 个碱基缺失所致。此外,在 S1 N 端区域积累了最多数量的氨基酸变异,导致分离株存在高变异性。S 蛋白肽水平的序列比较表明,7 株韩国分离株彼此之间的相似性最高可达 99.6%,最低可达 93.6%,但与其他参考株的相似性为 92.2%至 93.7%。总的来说,序列分析数据表明,目前在韩国流行的 PEDV 分离株具有多样性,代表了一个异质群体。系统进化分析显示出两个独立的聚类,所有韩国田间分离株都聚集在第二个聚类(聚类 2)中。结果表明,在韩国流行的分离株在系统进化上彼此之间的关系更为密切,而不是与其他参考株。有趣的是,基于代表 S1 结构域或 S1 N 端区域的核苷酸序列构建的系统进化树拓扑结构最接近基于全长 S 基因构建的系统进化树。因此,我们的数据表明,包括 N 端区域在内的部分 S 蛋白基因可能有助于揭示 PEDV 分离株的遗传相关性。