Howard Hughes Medical Institute, Janelia Farm Research Campus, Ashburn, Virginia, USA.
PLoS Comput Biol. 2010 Feb 5;6(2):e1000668. doi: 10.1371/journal.pcbi.1000668.
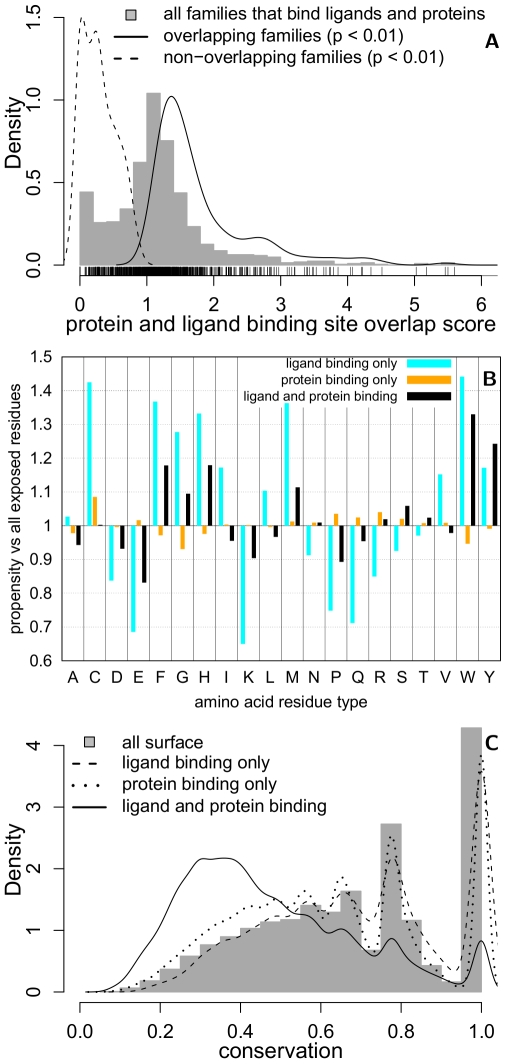
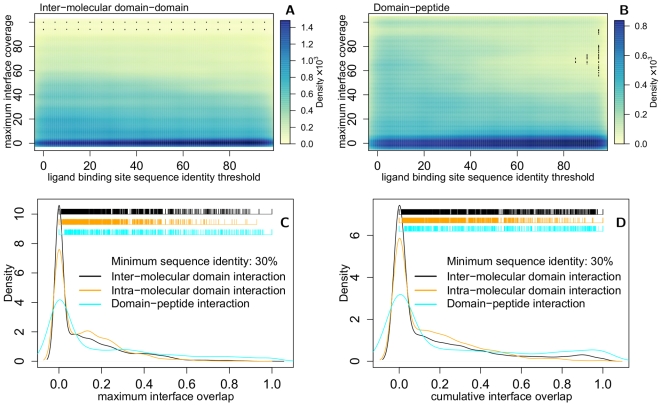
Protein-protein interactions are challenging targets for modulation by small molecules. Here, we propose an approach that harnesses the increasing structural coverage of protein complexes to identify small molecules that may target protein interactions. Specifically, we identify ligand and protein binding sites that overlap upon alignment of homologous proteins. Of the 2,619 protein structure families observed to bind proteins, 1,028 also bind small molecules (250-1000 Da), and 197 exhibit a statistically significant (p<0.01) overlap between ligand and protein binding positions. These "bi-functional positions", which bind both ligands and proteins, are particularly enriched in tyrosine and tryptophan residues, similar to "energetic hotspots" described previously, and are significantly less conserved than mono-functional and solvent exposed positions. Homology transfer identifies ligands whose binding sites overlap at least 20% of the protein interface for 35% of domain-domain and 45% of domain-peptide mediated interactions. The analysis recovered known small-molecule modulators of protein interactions as well as predicted new interaction targets based on the sequence similarity of ligand binding sites. We illustrate the predictive utility of the method by suggesting structural mechanisms for the effects of sanglifehrin A on HIV virion production, bepridil on the cellular entry of anthrax edema factor, and fusicoccin on vertebrate developmental pathways. The results, available at http://pibase.janelia.org, represent a comprehensive collection of structurally characterized modulators of protein interactions, and suggest that homologous structures are a useful resource for the rational design of interaction modulators.
蛋白质-蛋白质相互作用是小分子调节的挑战性靶标。在这里,我们提出了一种利用蛋白质复合物结构覆盖度增加的方法来识别可能靶向蛋白质相互作用的小分子。具体来说,我们确定了在同源蛋白质对齐时重叠的配体和蛋白质结合位点。在所观察到的 2619 个结合蛋白质的蛋白质结构家族中,有 1028 个也结合小分子(250-1000 Da),并且有 197 个表现出配体和蛋白质结合位置之间存在统计学上显著(p<0.01)的重叠。这些“双功能位置”既结合配体又结合蛋白质,特别富含酪氨酸和色氨酸残基,类似于以前描述的“能量热点”,并且比单功能和溶剂暴露位置保守性差。同源转移确定了结合位点与蛋白质界面重叠至少 20%的配体,对于 35%的域-域和 45%的域-肽介导相互作用。该分析根据配体结合位点的序列相似性,恢复了已知的蛋白质相互作用小分子调节剂以及预测的新相互作用靶标。我们通过建议结构机制来展示该方法的预测效用,这些结构机制涉及 sanglifehrin A 对 HIV 病毒颗粒产生的影响、bepridil 对炭疽水肿因子细胞进入的影响以及 fusicoccin 对脊椎动物发育途径的影响。这些结果可在 http://pibase.janelia.org 上获得,代表了结构上表征的蛋白质相互作用调节剂的综合集合,并表明同源结构是合理设计相互作用调节剂的有用资源。