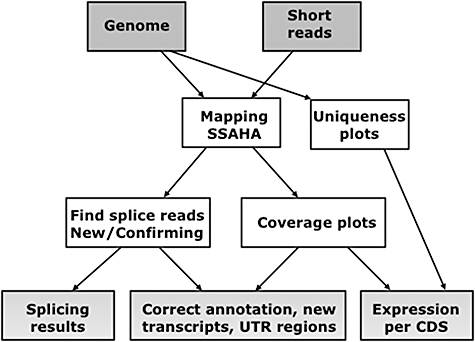
Parasite Genomics, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK.
Mol Microbiol. 2010 Apr;76(1):12-24. doi: 10.1111/j.1365-2958.2009.07026.x. Epub 2010 Feb 4.
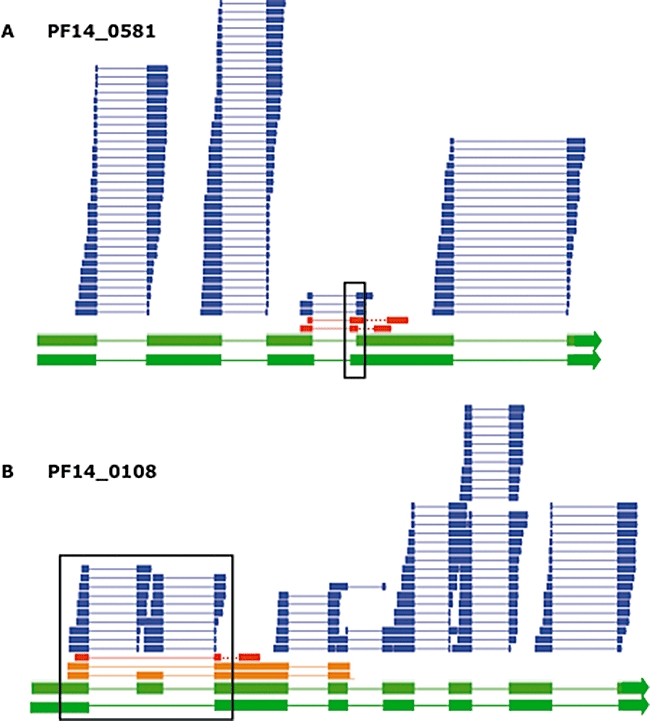
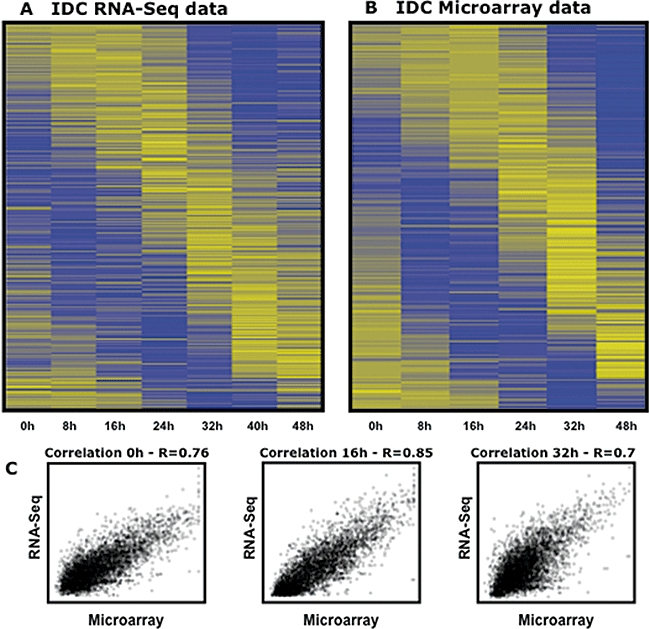
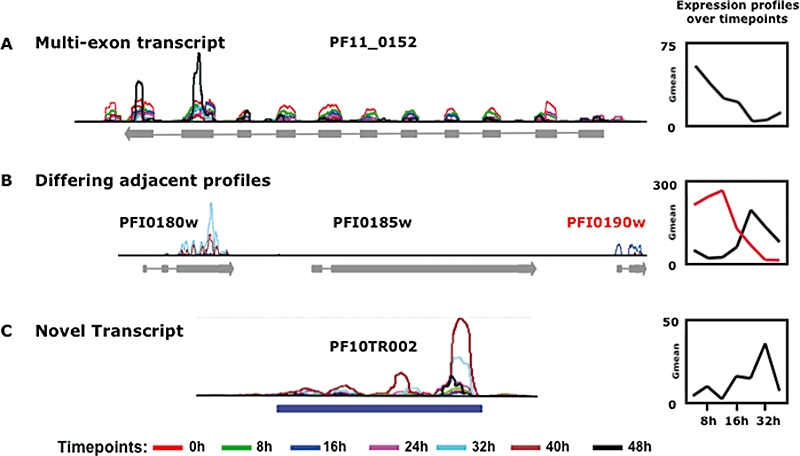
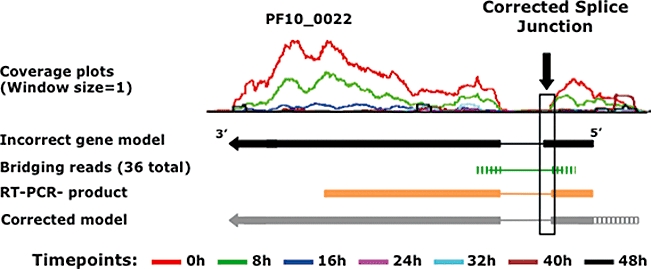
Recent advances in high-throughput sequencing present a new opportunity to deeply probe an organism's transcriptome. In this study, we used Illumina-based massively parallel sequencing to gain new insight into the transcriptome (RNA-Seq) of the human malaria parasite, Plasmodium falciparum. Using data collected at seven time points during the intraerythrocytic developmental cycle, we (i) detect novel gene transcripts; (ii) correct hundreds of gene models; (iii) propose alternative splicing events; and (iv) predict 5' and 3' untranslated regions. Approximately 70% of the unique sequencing reads map to previously annotated protein-coding genes. The RNA-Seq results greatly improve existing annotation of the P. falciparum genome with over 10% of gene models modified. Our data confirm 75% of predicted splice sites and identify 202 new splice sites, including 84 previously uncharacterized alternative splicing events. We also discovered 107 novel transcripts and expression of 38 pseudogenes, with many demonstrating differential expression across the developmental time series. Our RNA-Seq results correlate well with DNA microarray analysis performed in parallel on the same samples, and provide improved resolution over the microarray-based method. These data reveal new features of the P. falciparum transcriptional landscape and significantly advance our understanding of the parasite's red blood cell-stage transcriptome.
高通量测序的最新进展为深入研究生物体的转录组提供了新的机会。在这项研究中,我们使用基于 Illumina 的大规模平行测序技术,深入了解人类疟原虫(Plasmodium falciparum)的转录组(RNA-Seq)。我们利用在红细胞内发育周期的七个时间点收集的数据,(i)检测新的基因转录本;(ii)纠正数百个基因模型;(iii)提出可变剪接事件;以及(iv)预测 5' 和 3' 非翻译区。大约 70%的独特测序读数映射到先前注释的编码蛋白质的基因。RNA-Seq 结果极大地改进了现有的 P. falciparum 基因组注释,超过 10%的基因模型被修改。我们的数据证实了 75%的预测剪接位点,并确定了 202 个新的剪接位点,包括 84 个以前未表征的可变剪接事件。我们还发现了 107 个新的转录本和 38 个假基因的表达,其中许多在整个发育时间序列中表现出差异表达。我们的 RNA-Seq 结果与在相同样本上同时进行的 DNA 微阵列分析高度相关,并提供了比基于微阵列的方法更高的分辨率。这些数据揭示了 P. falciparum 转录景观的新特征,并大大提高了我们对寄生虫红细胞阶段转录组的理解。