Department of Medical Informatics, Biometry and Epidemiology, University of Munich, Marchioninistr 15, D-81377 Munich, Germany.
BMC Bioinformatics. 2010 Feb 8;11:78. doi: 10.1186/1471-2105-11-78.
While high-dimensional molecular data such as microarray gene expression data have been used for disease outcome prediction or diagnosis purposes for about ten years in biomedical research, the question of the additional predictive value of such data given that classical predictors are already available has long been under-considered in the bioinformatics literature.
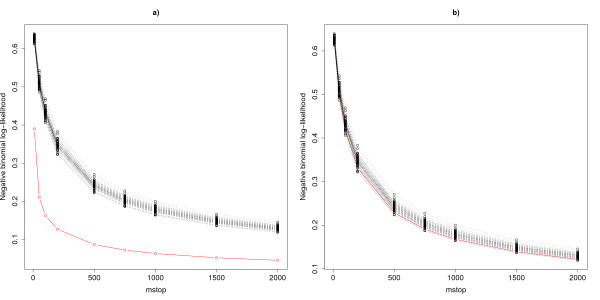
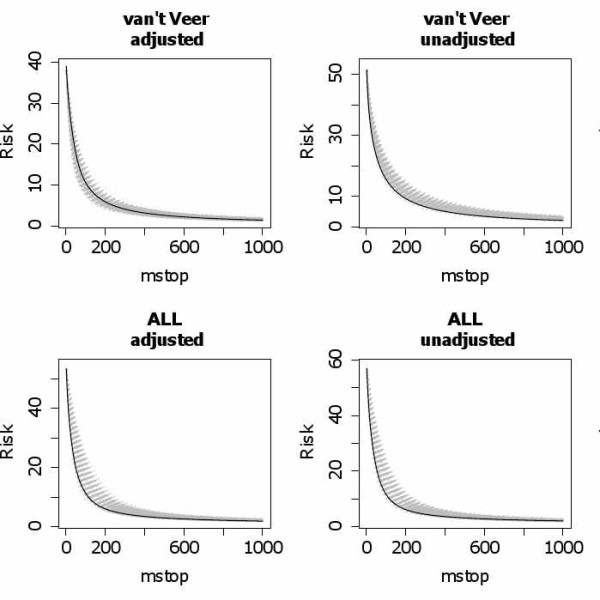
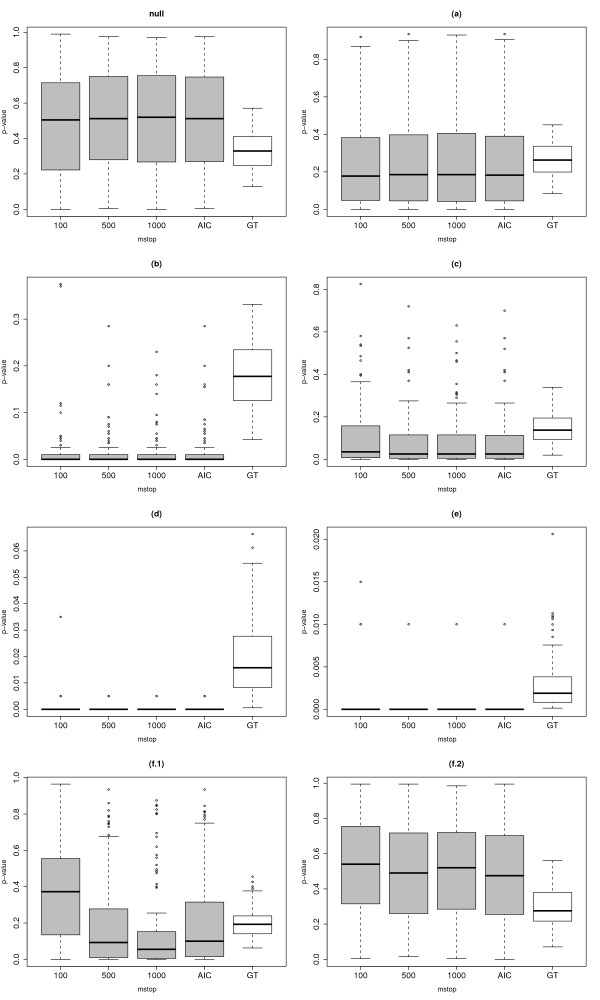
We suggest an intuitive permutation-based testing procedure for assessing the additional predictive value of high-dimensional molecular data. Our method combines two well-known statistical tools: logistic regression and boosting regression. We give clear advice for the choice of the only method parameter (the number of boosting iterations). In simulations, our novel approach is found to have very good power in different settings, e.g. few strong predictors or many weak predictors. For illustrative purpose, it is applied to the two publicly available cancer data sets.
Our simple and computationally efficient approach can be used to globally assess the additional predictive power of a large number of candidate predictors given that a few clinical covariates or a known prognostic index are already available. It is implemented in the R package "globalboosttest" which is publicly available from R-forge and will be sent to the CRAN as soon as possible.
尽管高维分子数据(如微阵列基因表达数据)在生物医学研究中已经用于疾病预后预测或诊断目的约十年,但在生物信息学文献中,对于给定已经存在的经典预测因子,此类数据的额外预测价值的问题长期以来一直被忽视。
我们建议了一种直观的基于置换的测试程序,用于评估高维分子数据的额外预测价值。我们的方法结合了两个著名的统计工具:逻辑回归和提升回归。我们为唯一的方法参数(提升迭代次数)提供了明确的选择建议。在模拟中,我们的新方法在不同的设置中具有很好的功效,例如少数强预测因子或许多弱预测因子。为了说明目的,我们将其应用于两个公开的癌症数据集。
我们的简单且计算效率高的方法可用于在已经存在少数临床协变量或已知预后指数的情况下,全局评估大量候选预测因子的额外预测能力。它在 R 包“globalboosttest”中实现,该包可从 R-forge 获得,并将尽快发送到 CRAN。