Department of Agricultural Biotechnology, Center for Fungal Pathogenesis, Center for Agricultural Biomaterials and Center for Fungal Genetic Resources, Seoul National University, Seoul 151-921, Korea.
BMC Genomics. 2010 Feb 10;11:104. doi: 10.1186/1471-2164-11-104.
Infection of plants by pathogens and the subsequent disease development involves substantial changes in the biochemistry and physiology of both partners. Analysis of genes that are expressed during these interactions represents a powerful strategy to obtain insights into the molecular events underlying these changes. We have employed expressed sequence tag (EST) analysis to identify rice genes involved in defense responses against infection by the blast fungus Magnaporthe oryzae and fungal genes involved in infectious growth within the host during a compatible interaction.
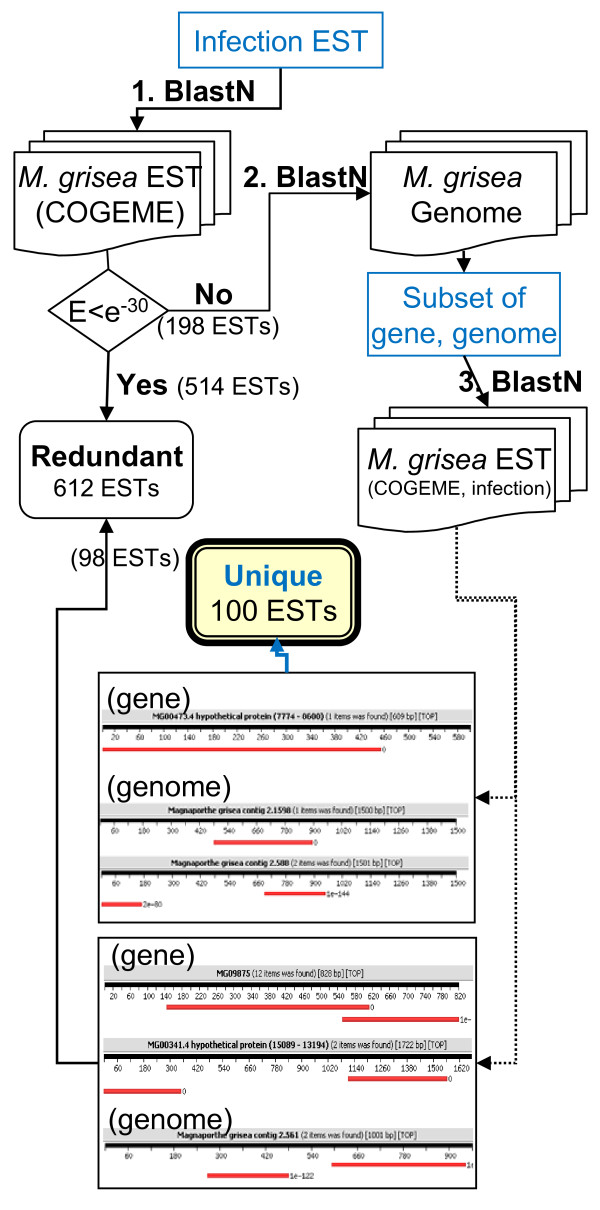
A cDNA library was constructed with RNA from rice leaves (Oryza sativa cv. Hwacheong) infected with M. oryzae strain KJ201. To enrich for fungal genes, subtraction library using PCR-based suppression subtractive hybridization was constructed with RNA from infected rice leaves as a tester and that from uninfected rice leaves as the driver. A total of 4,148 clones from two libraries were sequenced to generate 2,302 non-redundant ESTs. Of these, 712 and 1,562 ESTs could be identified to encode fungal and rice genes, respectively. To predict gene function, Gene Ontology (GO) analysis was applied, with 31% and 32% of rice and fungal ESTs being assigned to GO terms, respectively. One hundred uniESTs were found to be specific to fungal infection EST. More than 80 full-length fungal cDNA sequences were used to validate ab initio annotated gene model of M. oryzae genome sequence.
This study shows the power of ESTs to refine genome annotation and functional characterization. Results of this work have advanced our understanding of the molecular mechanisms underpinning fungal-plant interactions and formed the basis for new hypothesis.
植物受到病原体感染以及随后的发病过程涉及到两个伙伴的生物化学和生理学的重大变化。分析在这些相互作用过程中表达的基因是深入了解这些变化背后的分子事件的有力策略。我们采用表达序列标签(EST)分析来鉴定水稻中参与防御反应的基因,这些基因可抵抗稻瘟病菌(Magnaporthe oryzae)的感染,还鉴定了真菌中参与与宿主互作过程中感染性生长的基因。
我们构建了一个 cDNA 文库,该文库的 RNA 来源于被稻瘟病菌(M. oryzae 菌株 KJ201)感染的水稻叶片(Hwacheong 品种)。为了富集真菌基因,我们以感染水稻叶片的 RNA 为 tester,未感染水稻叶片的 RNA 为 driver,构建了基于 PCR 的抑制性消减杂交的消减文库。从两个文库中共测序了 4148 个克隆,生成了 2302 个非冗余 EST。其中,712 个和 1562 个 EST 分别可以被鉴定为编码真菌和水稻基因。为了预测基因功能,我们进行了基因本体论(GO)分析,结果显示 31%的水稻 EST 和 32%的真菌 EST 分别被分配到 GO 术语中。100 个 uniEST 被鉴定为专一地与真菌感染相关。使用 80 多个全长真菌 cDNA 序列验证了稻瘟病菌基因组序列的从头注释基因模型。
本研究表明 EST 对于完善基因组注释和功能特性分析非常有效。这项工作的结果增进了我们对真菌-植物相互作用分子机制的理解,并为新的假说奠定了基础。