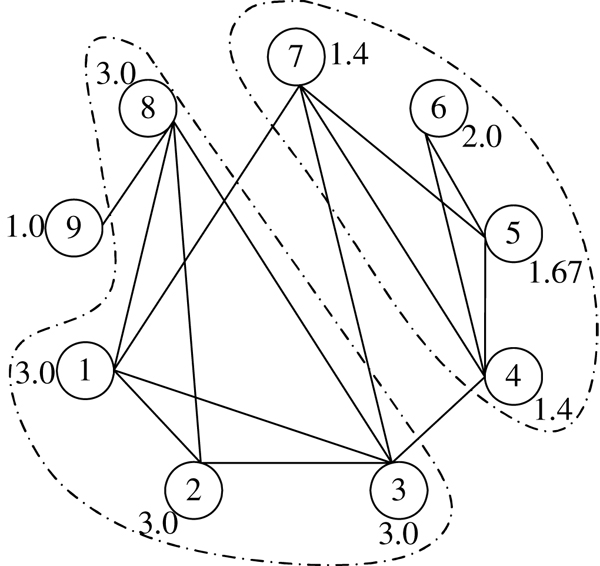
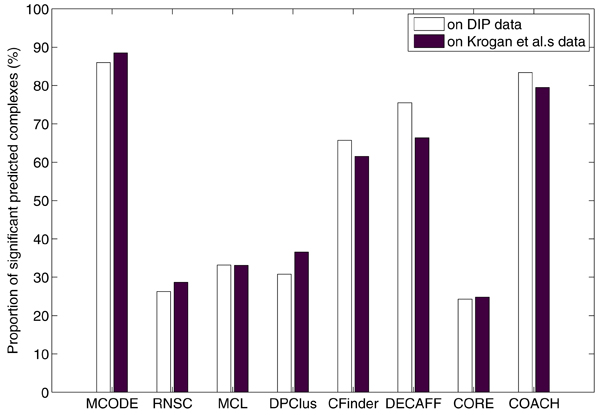
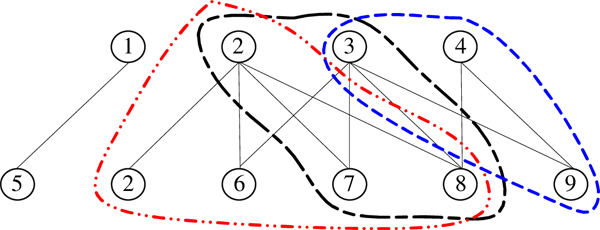
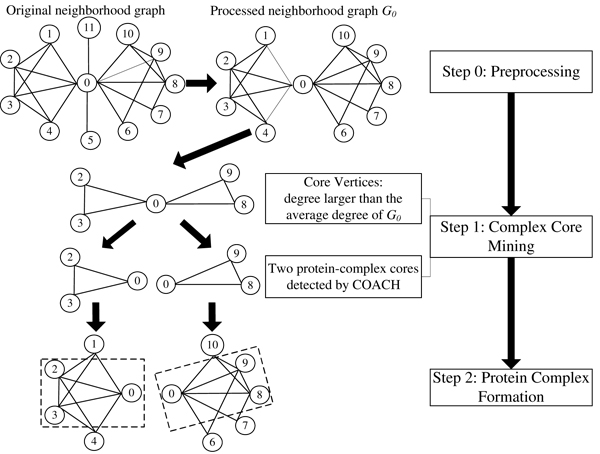
Institute for Infocomm Research, 1 Fusionopolis Way, Singapore.
BMC Genomics. 2010 Feb 10;11 Suppl 1(Suppl 1):S3. doi: 10.1186/1471-2164-11-S1-S3.
Most proteins form macromolecular complexes to perform their biological functions. However, experimentally determined protein complex data, especially of those involving more than two protein partners, are relatively limited in the current state-of-the-art high-throughput experimental techniques. Nevertheless, many techniques (such as yeast-two-hybrid) have enabled systematic screening of pairwise protein-protein interactions en masse. Thus computational approaches for detecting protein complexes from protein interaction data are useful complements to the limited experimental methods. They can be used together with the experimental methods for mapping the interactions of proteins to understand how different proteins are organized into higher-level substructures to perform various cellular functions.
Given the abundance of pairwise protein interaction data from high-throughput genome-wide experimental screenings, a protein interaction network can be constructed from protein interaction data by considering individual proteins as the nodes, and the existence of a physical interaction between a pair of proteins as a link. This binary protein interaction graph can then be used for detecting protein complexes using graph clustering techniques. In this paper, we review and evaluate the state-of-the-art techniques for computational detection of protein complexes, and discuss some promising research directions in this field.
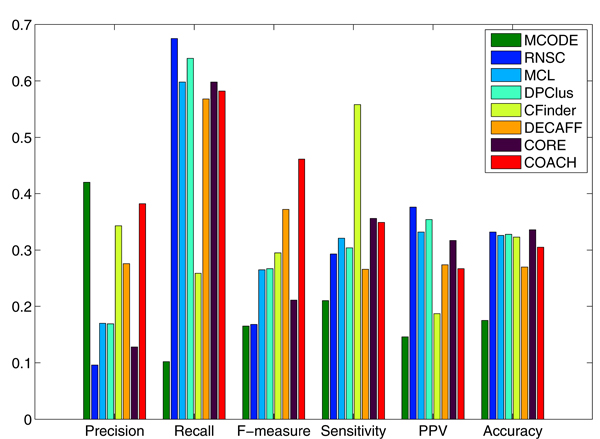
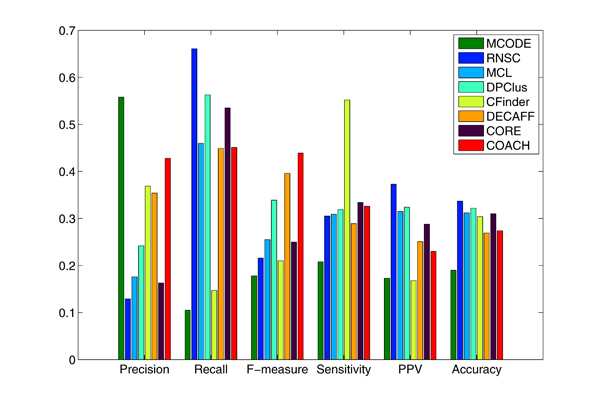
Experimental results with yeast protein interaction data show that the interaction subgraphs discovered by various computational methods matched well with actual protein complexes. In addition, the computational approaches have also improved in performance over the years. Further improvements could be achieved if the quality of the underlying protein interaction data can be considered adequately to minimize the undesirable effects from the irrelevant and noisy sources, and the various biological evidences can be better incorporated into the detection process to maximize the exploitation of the increasing wealth of biological knowledge available.
大多数蛋白质形成大分子复合物来执行其生物功能。然而,在当前的高通量实验技术中,实验确定的蛋白质复合物数据,尤其是涉及两个以上蛋白质伴侣的复合物数据相对有限。尽管如此,许多技术(如酵母双杂交)已经能够大规模系统地筛选成对的蛋白质-蛋白质相互作用。因此,从蛋白质相互作用数据中检测蛋白质复合物的计算方法是对有限的实验方法的有用补充。它们可以与实验方法一起用于绘制蛋白质相互作用,以了解不同的蛋白质如何组织成更高层次的亚结构来执行各种细胞功能。
鉴于高通量全基因组实验筛选获得的大量成对蛋白质相互作用数据,可以通过将单个蛋白质视为节点,将一对蛋白质之间存在物理相互作用视为链接,从蛋白质相互作用数据构建蛋白质相互作用网络。然后可以使用图聚类技术从二进制蛋白质相互作用图中检测蛋白质复合物。在本文中,我们回顾和评估了用于计算检测蛋白质复合物的最新技术,并讨论了该领域一些有前途的研究方向。
使用酵母蛋白质相互作用数据的实验结果表明,各种计算方法发现的相互作用子图与实际蛋白质复合物很好地匹配。此外,计算方法的性能在这些年来也得到了提高。如果能够充分考虑基础蛋白质相互作用数据的质量,以最小化来自不相关和嘈杂源的不良影响,并且能够更好地将各种生物学证据纳入检测过程中,以最大限度地利用现有的越来越多的生物学知识财富,那么还可以进一步提高。