Omranian Sara, Nikoloski Zoran, Grimm Dominik G
Technical University of Munich, Campus Straubing for Biotechnology and Sustainability, Bioinformatics, Petersgasse 18, 94315 Straubing, Germany.
Weihenstephan-Triesdorf University of Applied Sciences, Petersgasse 18, 94315 Straubing, Germany.
Comput Struct Biotechnol J. 2022 May 27;20:2699-2712. doi: 10.1016/j.csbj.2022.05.049. eCollection 2022.
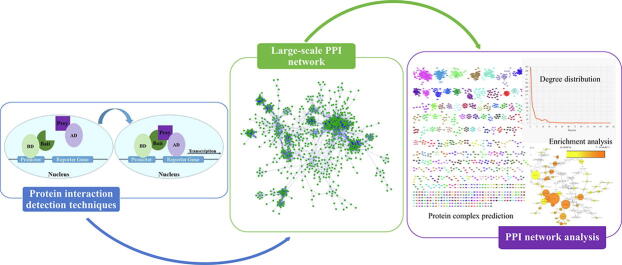
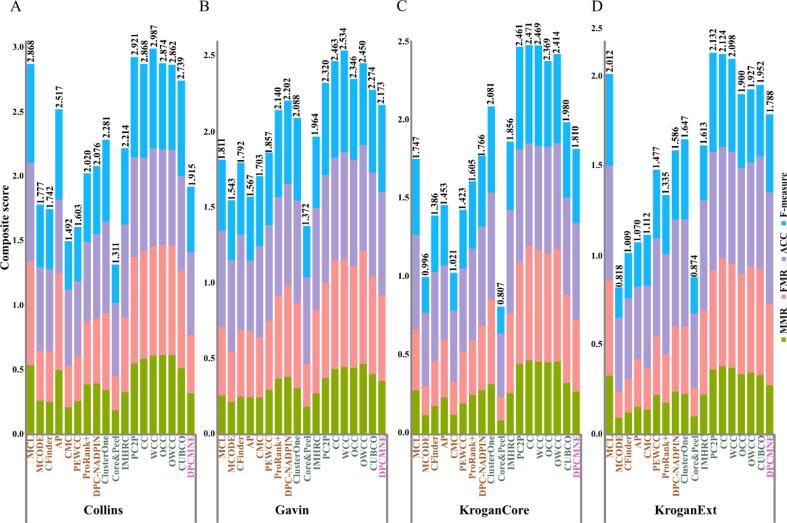
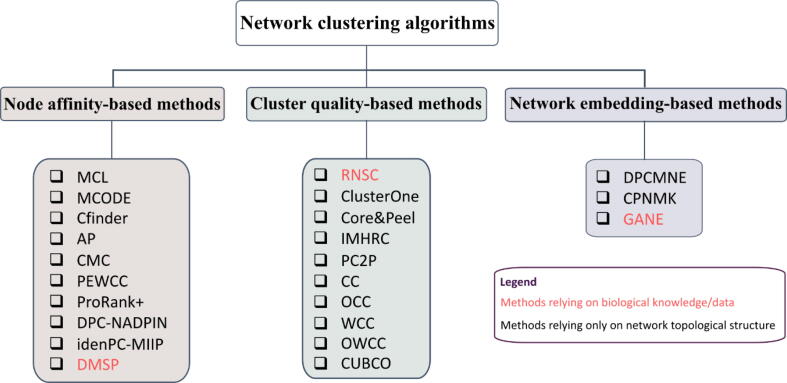
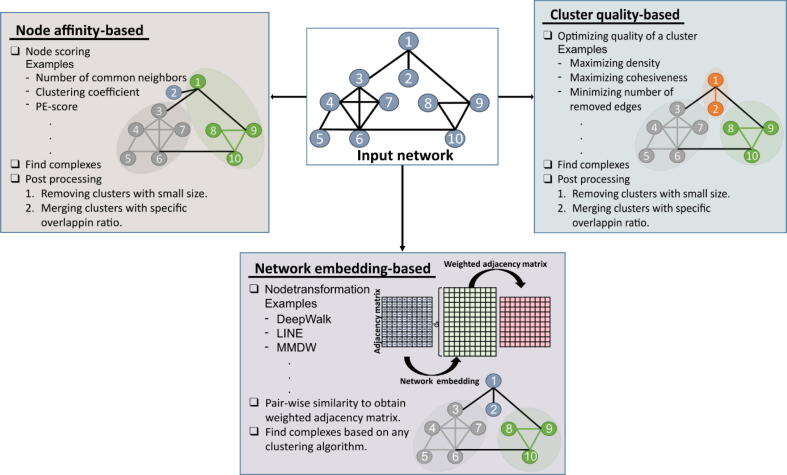
Physically interacting proteins form macromolecule complexes that drive diverse cellular processes. Advances in experimental techniques that capture interactions between proteins provide us with protein-protein interaction (PPI) networks from several model organisms. These datasets have enabled the prediction and other computational analyses of protein complexes. Here we provide a systematic review of the state-of-the-art algorithms for protein complex prediction from PPI networks proposed in the past two decades. The existing approaches that solve this problem are categorized into three groups, including: cluster-quality-based, node affinity-based, and network embedding-based approaches, and we compare and contrast the advantages and disadvantages. We further include a comparative analysis by computing the performance of eighteen methods based on twelve well-established performance measures on four widely used benchmark protein-protein interaction networks. Finally, the limitations and drawbacks of both, current data and approaches, along with the potential solutions in this field are discussed, with emphasis on the points that pave the way for future research efforts in this field.
物理上相互作用的蛋白质形成驱动各种细胞过程的大分子复合物。捕获蛋白质之间相互作用的实验技术进展为我们提供了来自几种模式生物的蛋白质-蛋白质相互作用(PPI)网络。这些数据集使得蛋白质复合物的预测和其他计算分析成为可能。在此,我们对过去二十年中提出的用于从PPI网络预测蛋白质复合物的最先进算法进行了系统综述。解决该问题的现有方法分为三类,包括:基于聚类质量的方法、基于节点亲和力的方法和基于网络嵌入的方法,我们对其优缺点进行了比较和对比。我们还基于四个广泛使用的基准蛋白质-蛋白质相互作用网络,通过计算十八种方法在十二种成熟性能指标上的性能进行了比较分析。最后,讨论了当前数据和方法的局限性和缺点,以及该领域的潜在解决方案,重点强调了为该领域未来研究工作铺平道路的要点。