Hubrecht Institute and University Medical Center Utrecht, KNAW, Uppsalalaan 8, 3584 CT Utrecht, The Netherlands.
Nucleic Acids Res. 2010 Jun;38(10):e116. doi: 10.1093/nar/gkq072. Epub 2010 Feb 17.
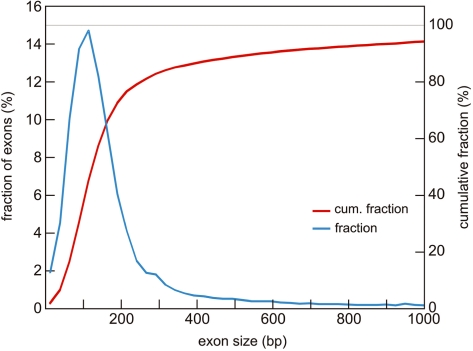
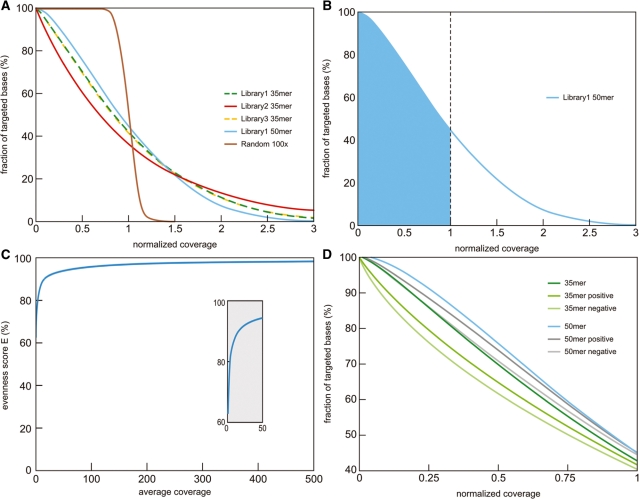
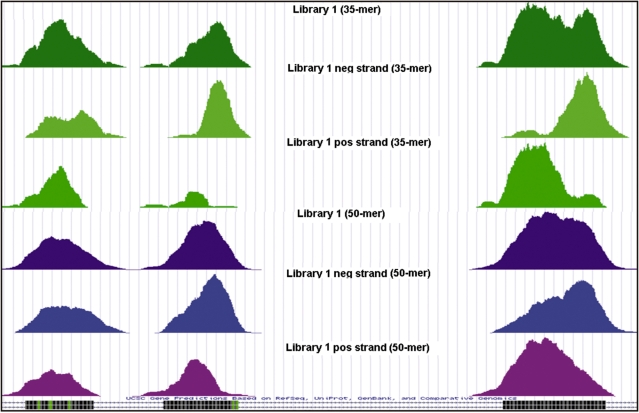
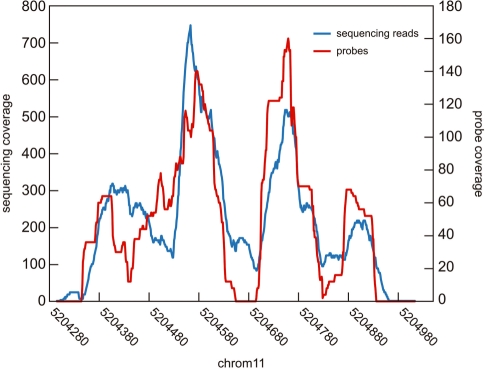
Microarray-based enrichment of selected genomic loci is a powerful method for genome complexity reduction for next-generation sequencing. Since the vast majority of exons in vertebrate genomes are smaller than 150 nt, we explored the use of short fragment libraries (85-110 bp) to achieve higher enrichment specificity by reducing carryover and adverse effects of flanking intronic sequences. High enrichment specificity (60-75%) was obtained with a relative even base coverage. Up to 98% of the target-sequence was covered more than 20x at an average coverage depth of about 200x. To verify the accuracy of SNP/mutation detection, we evaluated 384 known non-reference SNPs in the targeted regions. At approximately 200x average sequence coverage, we were able to survey 96.4% of 1.69 Mb of genomic sequence with only 4.2% false negative calls, mostly due to low coverage. Using the same settings, a total of 1197 novel candidate variants were detected. Verification experiments revealed only eight false positive calls, indicating an overall false positive rate of less than 1 per approximately 200,000 bp. Taken together, short fragment libraries provide highly efficient and flexible enrichment of exonic targets and yield relatively even base coverage, which facilitates accurate SNP and mutation detection. Raw sequencing data, alignment files and called SNPs have been submitted into GEO database http://www.ncbi.nlm.nih.gov/geo/ with accession number GSE18542.
基于微阵列的选定基因组区域富集是降低下一代测序基因组复杂性的一种强大方法。由于脊椎动物基因组中绝大多数外显子都小于 150 nt,我们探索了使用短片段文库(85-110 bp)的方法,通过减少侧翼内含子序列的携带和不利影响来实现更高的富集特异性。通过相对均匀的碱基覆盖,获得了 60-75%的高富集特异性。高达 98%的目标序列在平均覆盖深度约为 200x 时被覆盖了 20x 以上。为了验证 SNP/突变检测的准确性,我们评估了靶向区域中的 384 个已知非参考 SNP。在大约 200x 的平均序列覆盖度下,我们能够在仅 4.2%的假阴性率下,对 1.69 Mb 的基因组序列进行调查,主要是由于覆盖度低。使用相同的设置,总共检测到 1197 个新的候选变体。验证实验仅显示了 8 个假阳性,表明总体假阳性率低于每 200,000 bp 约 1 个。总之,短片段文库可高效、灵活地富集外显子靶标,并产生相对均匀的碱基覆盖度,从而有助于 SNP 和突变的准确检测。原始测序数据、比对文件和已调用的 SNP 已提交到 GEO 数据库(http://www.ncbi.nlm.nih.gov/geo/), accession number 为 GSE18542。