Genetics and Population Health Division, Queensland Institute of Medical Research, Brisbane, Australia.
PLoS Genet. 2010 Feb 19;6(2):e1000850. doi: 10.1371/journal.pgen.1000850.
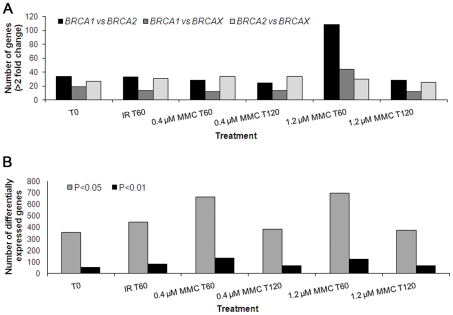
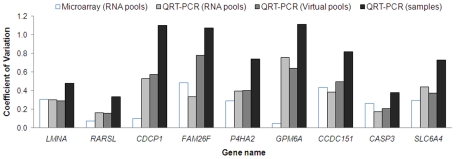
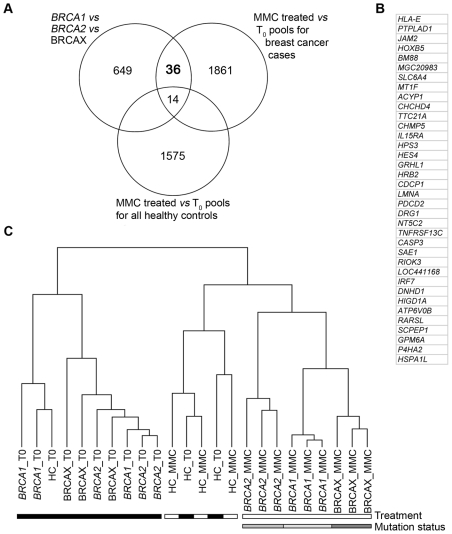
A large number of rare sequence variants of unknown clinical significance have been identified in the breast cancer susceptibility genes, BRCA1 and BRCA2. Laboratory-based methods that can distinguish between carriers of pathogenic mutations and non-carriers are likely to have utility for the classification of these sequence variants. To identify predictors of pathogenic mutation status in familial breast cancer patients, we explored the use of gene expression arrays to assess the effect of two DNA-damaging agents (irradiation and mitomycin C) on cellular response in relation to BRCA1 and BRCA2 mutation status. A range of regimes was used to treat 27 lymphoblastoid cell-lines (LCLs) derived from affected women in high-risk breast cancer families (nine BRCA1, nine BRCA2, and nine non-BRCA1/2 or BRCAX individuals) and nine LCLs from healthy individuals. Using an RNA-pooling strategy, we found that treating LCLs with 1.2 microM mitomycin C and measuring the gene expression profiles 1 hour post-treatment had the greatest potential to discriminate BRCA1, BRCA2, and BRCAX mutation status. A classifier was built using the expression profile of nine QRT-PCR validated genes that were associated with BRCA1, BRCA2, and BRCAX status in RNA pools. These nine genes could distinguish BRCA1 from BRCA2 carriers with 83% accuracy in individual samples, but three-way analysis for BRCA1, BRCA2, and BRCAX had a maximum of 59% prediction accuracy. Our results suggest that, compared to BRCA1 and BRCA2 mutation carriers, non-BRCA1/2 (BRCAX) individuals are genetically heterogeneous. This study also demonstrates the effectiveness of RNA pools to compare the expression profiles of cell-lines from BRCA1, BRCA2, and BRCAX cases after treatment with irradiation and mitomycin C as a method to prioritize treatment regimes for detailed downstream expression analysis.
大量具有未知临床意义的罕见序列变异已在乳腺癌易感基因 BRCA1 和 BRCA2 中被发现。能够区分致病性突变携带者和非携带者的基于实验室的方法可能对这些序列变异的分类有用。为了确定家族性乳腺癌患者中致病性突变状态的预测因子,我们探讨了使用基因表达谱来评估两种 DNA 损伤剂(辐射和丝裂霉素 C)对细胞反应的影响与 BRCA1 和 BRCA2 突变状态的关系。使用一系列方案来治疗 27 个来自高乳腺癌风险家族的受影响女性的淋巴母细胞系 (LCL)(9 个 BRCA1、9 个 BRCA2 和 9 个非 BRCA1/2 或 BRCAX 个体)和 9 个来自健康个体的 LCL。使用 RNA 池化策略,我们发现用 1.2 μM 丝裂霉素 C 处理 LCL 并在治疗后 1 小时测量基因表达谱最有可能区分 BRCA1、BRCA2 和 BRCAX 突变状态。使用与 RNA 池中的 BRCA1、BRCA2 和 BRCAX 状态相关的 9 个 qRT-PCR 验证基因的表达谱构建分类器。这九个基因可以在单个样本中以 83%的准确性区分 BRCA1 和 BRCA2 携带者,但对 BRCA1、BRCA2 和 BRCAX 的三向分析的预测准确率最高为 59%。我们的结果表明,与 BRCA1 和 BRCA2 突变携带者相比,非 BRCA1/2(BRCAX)个体在遗传上是异质的。本研究还证明了 RNA 池在比较经辐射和丝裂霉素 C 处理后的 BRCA1、BRCA2 和 BRCAX 病例的细胞系表达谱方面的有效性,作为一种方法来确定治疗方案的优先级,以便进行详细的下游表达分析。