Division of Biostatistics, Institute & Department of Public Health, National Yang-Ming University, Taipei 112, Taiwan.
BMC Bioinformatics. 2010 Feb 27;11:111. doi: 10.1186/1471-2105-11-111.
Combining data from different ethnic populations in a study can increase efficacy of methods designed to identify expression quantitative trait loci (eQTL) compared to analyzing each population independently. In such studies, however, the genetic diversity of minor allele frequencies among populations has rarely been taken into account. Due to the fact that allele frequency diversity and population-level expression differences are present in populations, a consensus regarding the optimal statistical approach for analysis of eQTL in data combining different populations remains inconclusive.
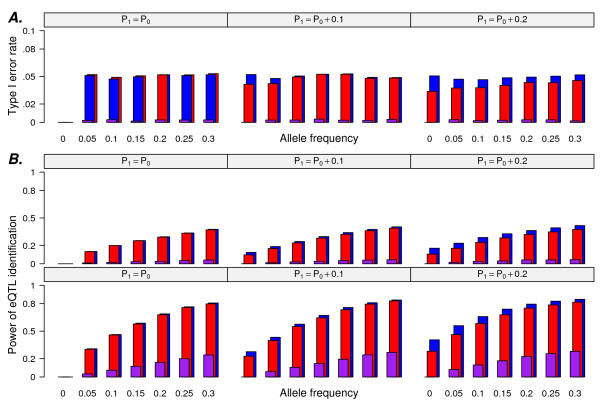
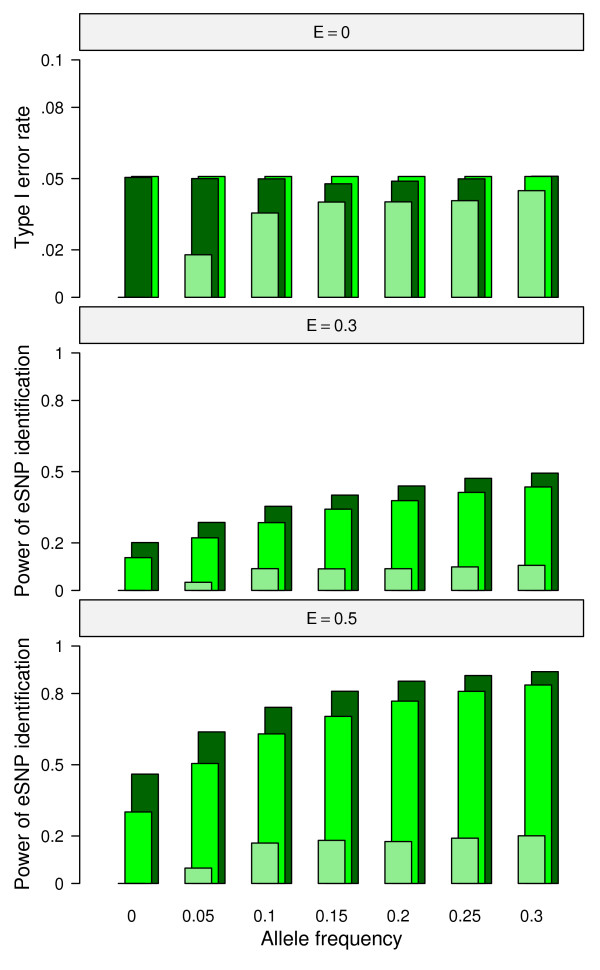
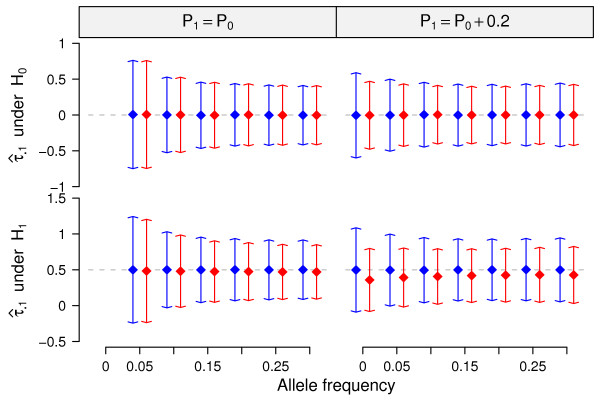
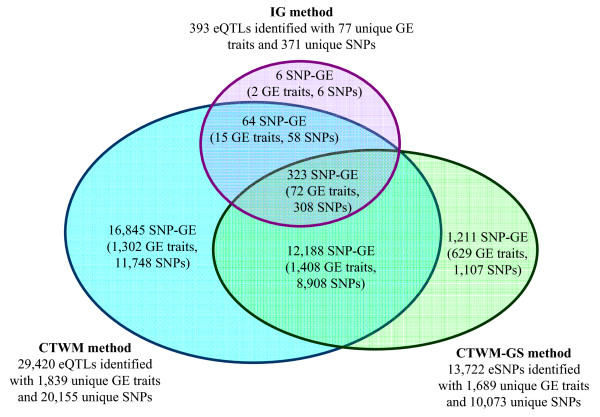
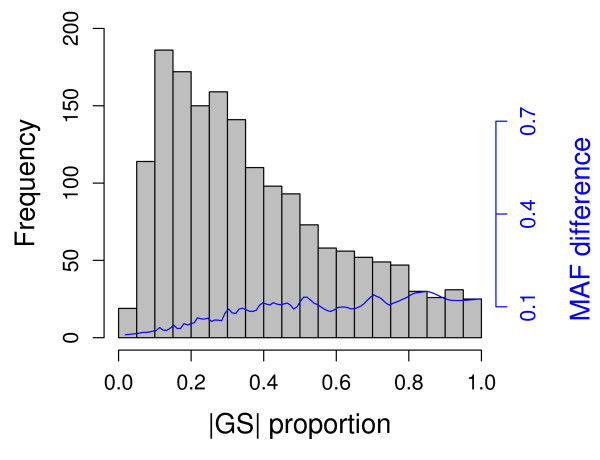
In this report, we explored the applicability of a constrained two-way model to identify eQTL for combined ethnic data that might contain genetic diversity among ethnic populations. In addition, gene expression differences resulted from ethnic allele frequency diversity between populations were directly estimated and analyzed by the constrained two-way model. Through simulation, we investigated effects of genetic diversity on eQTL identification by examining gene expression data pooled from normal quantile transformation of each population. Using the constrained two-way model to reanalyze data from Caucasians and Asian individuals available from HapMap, a large number of eQTL were identified with similar genetic effects on the gene expression levels in these two populations. Furthermore, 19 single nucleotide polymorphisms with inter-population differences with respect to both genotype frequency and gene expression levels directed by genotypes were identified and reflected a clear distinction between Caucasians and Asian individuals.
This study illustrates the influence of minor allele frequencies on common eQTL identification using either separate or combined population data. Our findings are important for future eQTL studies in which different datasets are combined to increase the power of eQTL identification.
与分别分析每个群体相比,在研究中将不同族群的数据结合起来可以提高用于识别表达数量性状基因座 (eQTL) 的方法的功效。然而,在这种研究中,很少考虑到群体之间的次要等位基因频率遗传多样性。由于在群体中存在等位基因频率多样性和群体水平的表达差异,因此对于分析不同群体数据结合的 eQTL 的最佳统计方法仍然没有达成共识。
在本报告中,我们探讨了受约束的双向模型在识别可能包含族群之间遗传多样性的混合族群数据的 eQTL 方面的适用性。此外,受约束的双向模型还直接估计和分析了由族群等位基因频率多样性引起的基因表达差异。通过模拟,我们通过检查从每个群体的正态分位数转换中汇总的基因表达数据,研究了遗传多样性对 eQTL 识别的影响。使用受约束的双向模型重新分析来自 HapMap 的白种人和亚洲个体的数据,在这两个群体中,大量的 eQTL 被鉴定出来,对基因表达水平具有相似的遗传影响。此外,鉴定了 19 个单核苷酸多态性,这些多态性在基因型频率和由基因型指导的基因表达水平方面在族群之间存在差异,并反映了白种人和亚洲个体之间的明显区别。
本研究说明了使用单独或混合群体数据识别常见 eQTL 时,次要等位基因频率的影响。我们的发现对于未来的 eQTL 研究很重要,这些研究将结合不同的数据集以提高 eQTL 识别的功效。