Ujino-Ihara Tokuko, Taguchi Yuriko, Moriguchi Yoshinari, Tsumura Yoshihiko
Department of Forest Genetics, Forestry and Forest Products Research Institute, Tsukuba, Ibaraki, 305-8687, Japan.
BMC Res Notes. 2010 Mar 2;3:51. doi: 10.1186/1756-0500-3-51.
In order to identify single nucleotide polymorphisms (SNPs) efficiently from a species with a large genome, SNPs were identified from an expressed sequence tag (EST) database combined with High Resolution Melting (HRM) analysis.
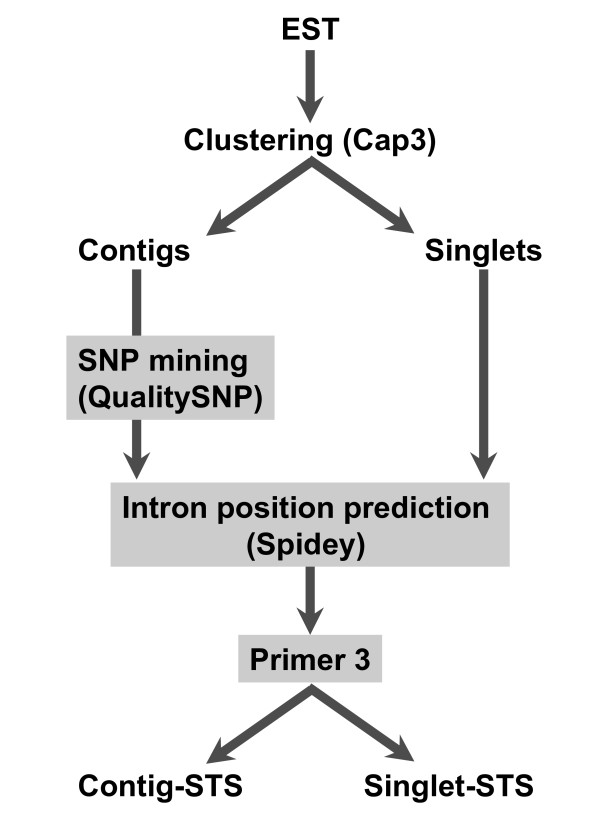
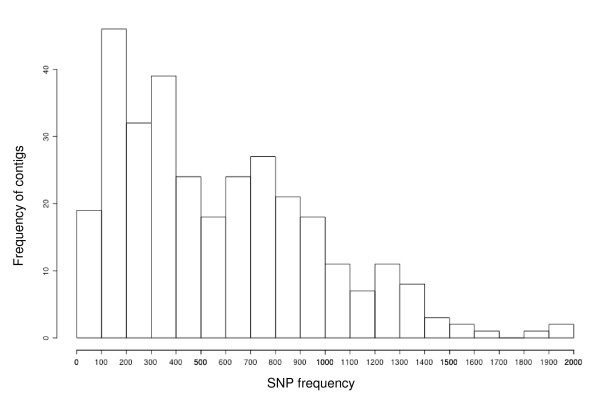
A total of 574 sequence tagged sites (STSs) were generated from Cryptomeria japonica and HRM analysis was used to screen for polymorphisms in these STS markers. STSs were designed in two ways: 1) putative SNP sites were identified by comparing ESTs from specific contigs, then 226 primer pairs designed for the purpose to amplify these SNPs; 2) 348 primer pairs were randomly designed using reads from the 3' end of cDNA. HRM analysis revealed that 325 markers among eight individuals were polymorphic, and that STSs, including putative SNP sites, exhibited higher levels of polymorphism.
Our results indicate that the combination of SNP screening from an EST database combined with HRM analysis is a highly efficient way to develop SNP markers for expressed genes. This method will contribute to both genetic mapping and the identification of SNPs in non-model organisms.
为了从具有大基因组的物种中高效鉴定单核苷酸多态性(SNP),结合高分辨率熔解曲线(HRM)分析,从表达序列标签(EST)数据库中鉴定SNP。
从日本柳杉中总共产生了574个序列标签位点(STS),并使用HRM分析筛选这些STS标记中的多态性。STS有两种设计方式:1)通过比较特定重叠群中的EST来鉴定推定的SNP位点,然后设计226对引物来扩增这些SNP;2)使用来自cDNA 3'端的读段随机设计348对引物。HRM分析表明,八个个体中的325个标记是多态性的,并且包括推定SNP位点的STS表现出更高水平的多态性。
我们的结果表明,从EST数据库中筛选SNP并结合HRM分析是开发表达基因SNP标记的高效方法。该方法将有助于非模式生物的遗传作图和SNP鉴定。