Center for Ecology and Evolutionary Biology, University of Oregon, Eugene, Oregon, United States of America.
PLoS Genet. 2010 Feb 26;6(2):e1000862. doi: 10.1371/journal.pgen.1000862.
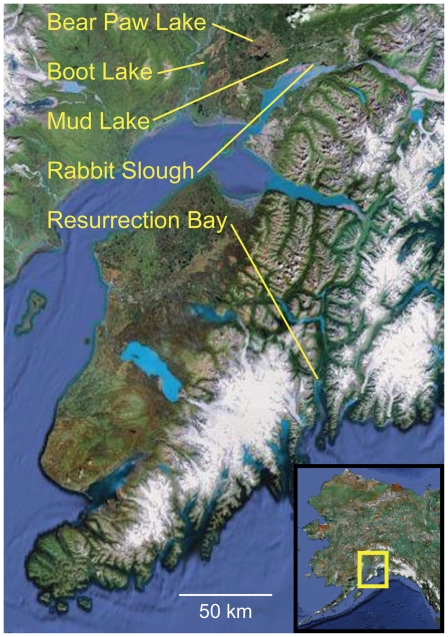
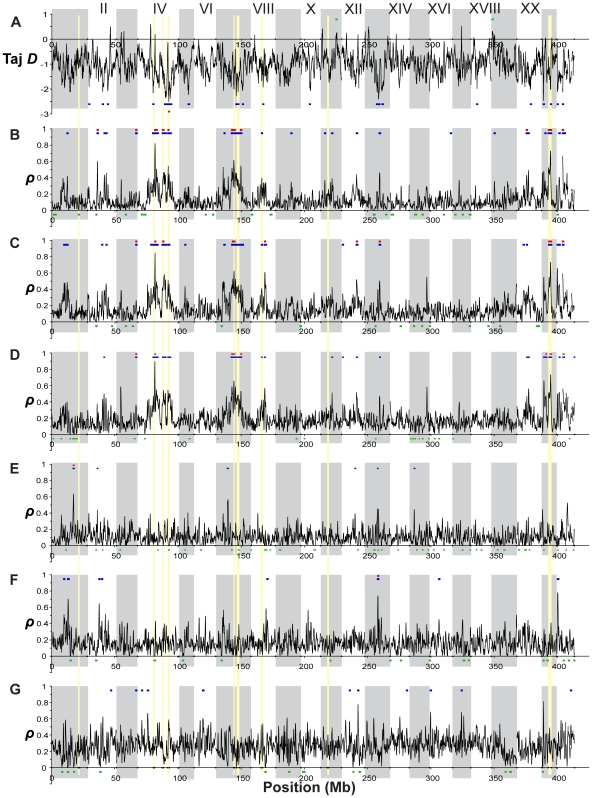
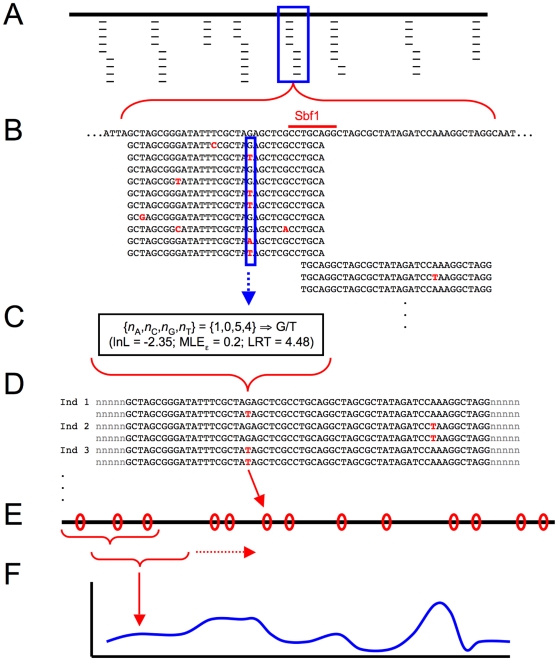
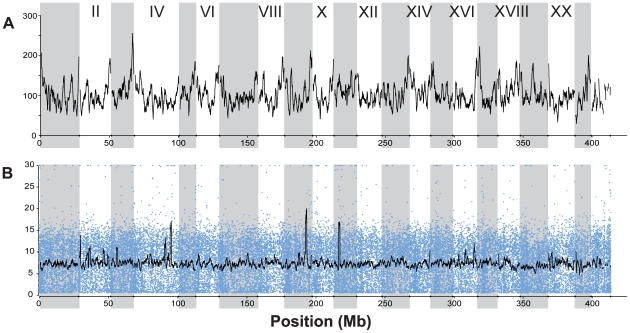
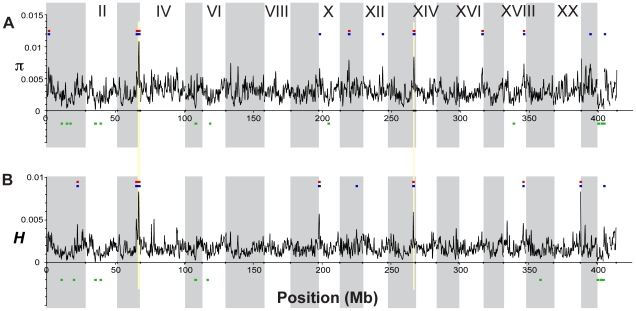
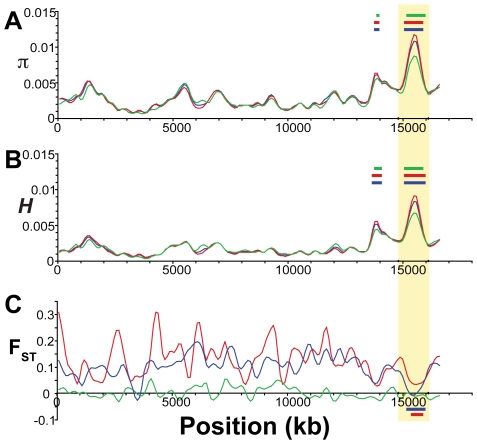
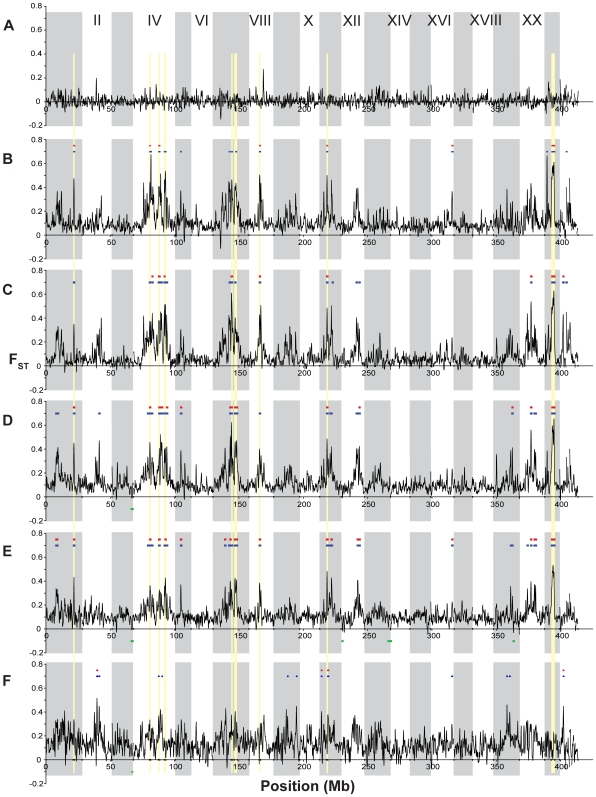
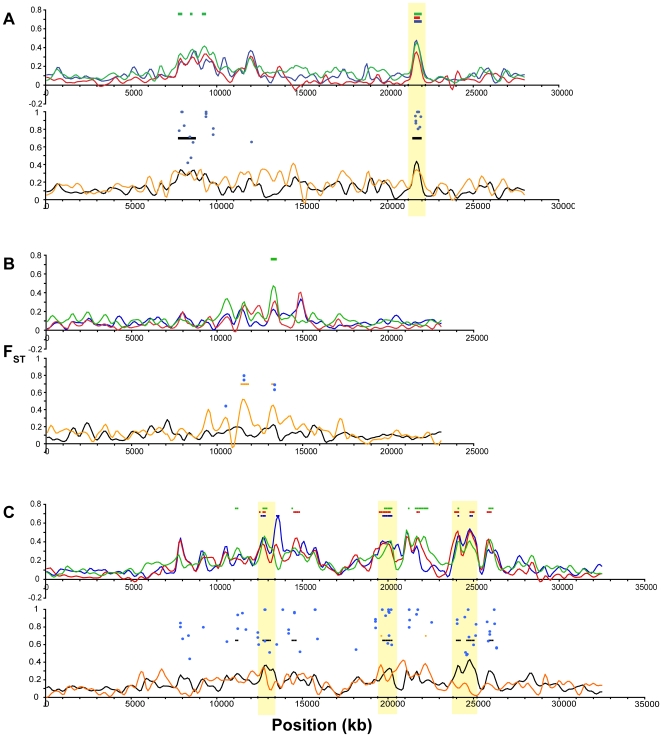
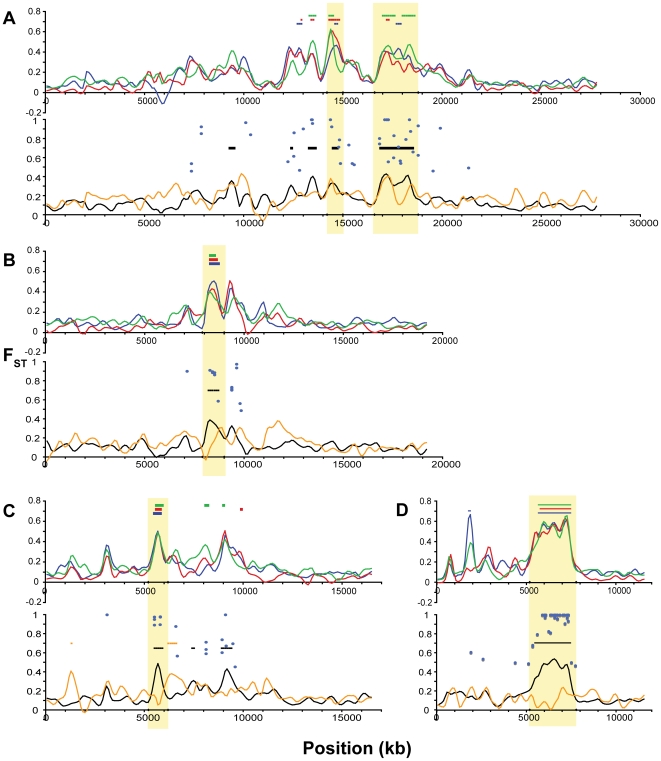
Next-generation sequencing technology provides novel opportunities for gathering genome-scale sequence data in natural populations, laying the empirical foundation for the evolving field of population genomics. Here we conducted a genome scan of nucleotide diversity and differentiation in natural populations of threespine stickleback (Gasterosteus aculeatus). We used Illumina-sequenced RAD tags to identify and type over 45,000 single nucleotide polymorphisms (SNPs) in each of 100 individuals from two oceanic and three freshwater populations. Overall estimates of genetic diversity and differentiation among populations confirm the biogeographic hypothesis that large panmictic oceanic populations have repeatedly given rise to phenotypically divergent freshwater populations. Genomic regions exhibiting signatures of both balancing and divergent selection were remarkably consistent across multiple, independently derived populations, indicating that replicate parallel phenotypic evolution in stickleback may be occurring through extensive, parallel genetic evolution at a genome-wide scale. Some of these genomic regions co-localize with previously identified QTL for stickleback phenotypic variation identified using laboratory mapping crosses. In addition, we have identified several novel regions showing parallel differentiation across independent populations. Annotation of these regions revealed numerous genes that are candidates for stickleback phenotypic evolution and will form the basis of future genetic analyses in this and other organisms. This study represents the first high-density SNP-based genome scan of genetic diversity and differentiation for populations of threespine stickleback in the wild. These data illustrate the complementary nature of laboratory crosses and population genomic scans by confirming the adaptive significance of previously identified genomic regions, elucidating the particular evolutionary and demographic history of such regions in natural populations, and identifying new genomic regions and candidate genes of evolutionary significance.
下一代测序技术为在自然种群中收集基因组规模的序列数据提供了新的机会,为不断发展的群体基因组学领域奠定了经验基础。在这里,我们对三刺鱼(Gasterosteus aculeatus)自然种群中的核苷酸多样性和分化进行了基因组扫描。我们使用 Illumina 测序的 RAD 标签,在来自两个海洋和三个淡水种群的 100 个个体中的每个个体中鉴定和分型了超过 45,000 个单核苷酸多态性(SNP)。种群间遗传多样性和分化的总体估计证实了生物地理学假说,即大型混合海洋种群多次产生表型上不同的淡水种群。表现出平衡和分歧选择特征的基因组区域在多个独立衍生的种群中非常一致,表明在刺鱼中复制的平行表型进化可能是通过在全基因组范围内广泛的平行遗传进化发生的。这些基因组区域中的一些与之前使用实验室图谱交叉识别的刺鱼表型变异的 QTL 区域重合。此外,我们还鉴定了一些在独立种群中表现出平行分化的新区域。这些区域的注释揭示了许多候选基因,它们是刺鱼表型进化的候选基因,将成为未来在该和其他生物体中进行遗传分析的基础。本研究代表了对野生三刺鱼种群进行的首次基于高密度 SNP 的遗传多样性和分化的基因组扫描。这些数据通过确认先前鉴定的基因组区域的适应性意义,阐明这些区域在自然种群中的特定进化和人口历史,并鉴定新的基因组区域和具有进化意义的候选基因,说明了实验室交叉和群体基因组扫描的互补性。