Faculty of Medicine and Health Sciences, United Arab Emirates University, Al-Ain, United Arab Emirates.
Hum Mol Genet. 2010 Jun 1;19(11):2239-50. doi: 10.1093/hmg/ddq103. Epub 2010 Mar 10.
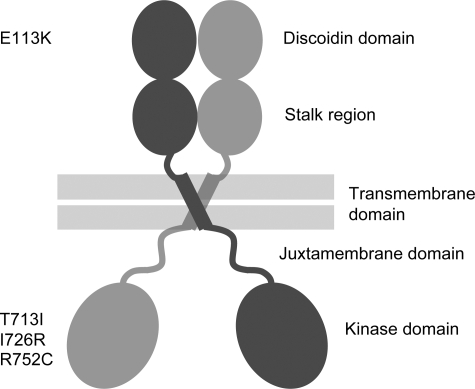
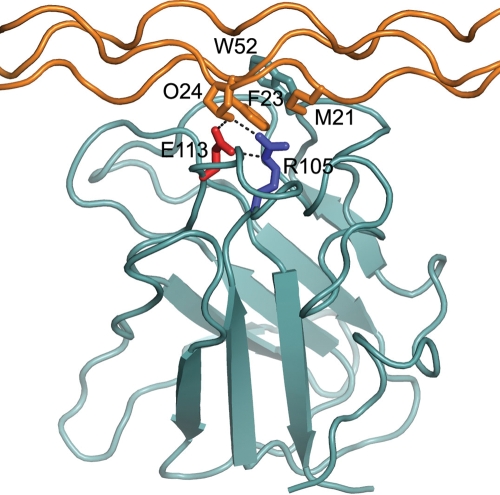
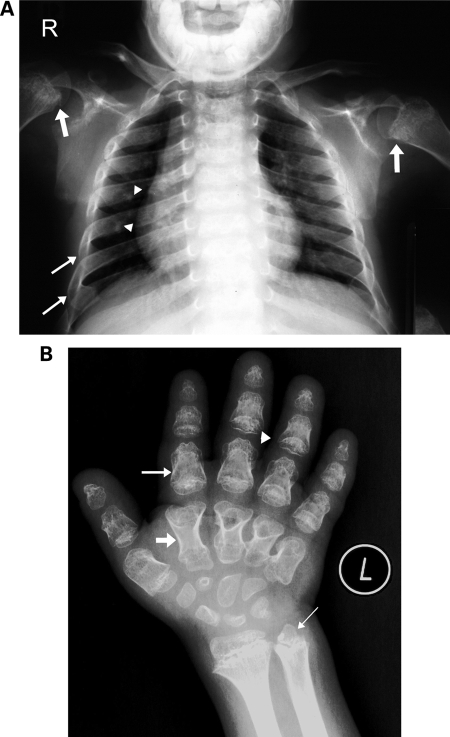
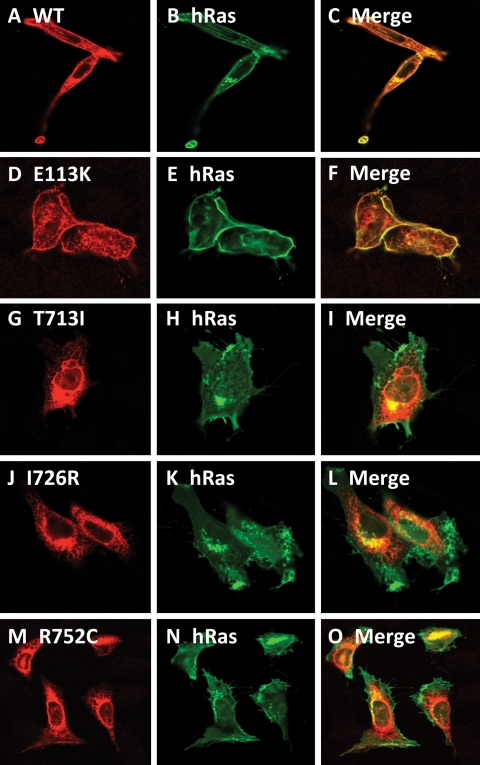
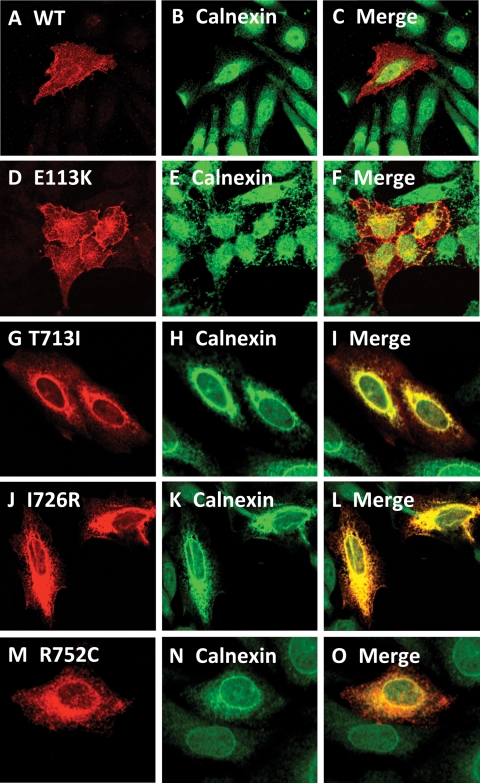
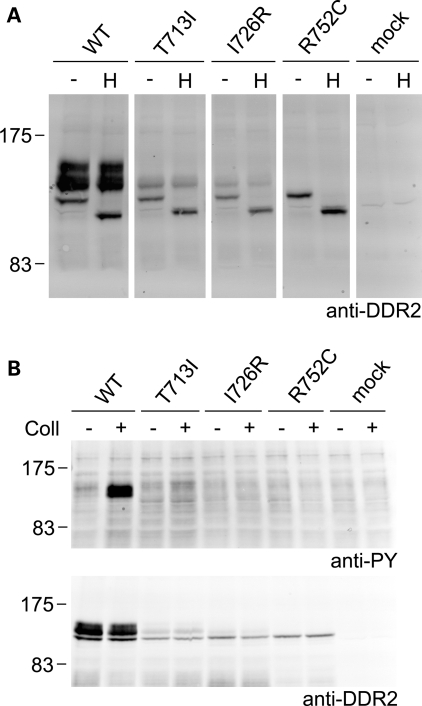
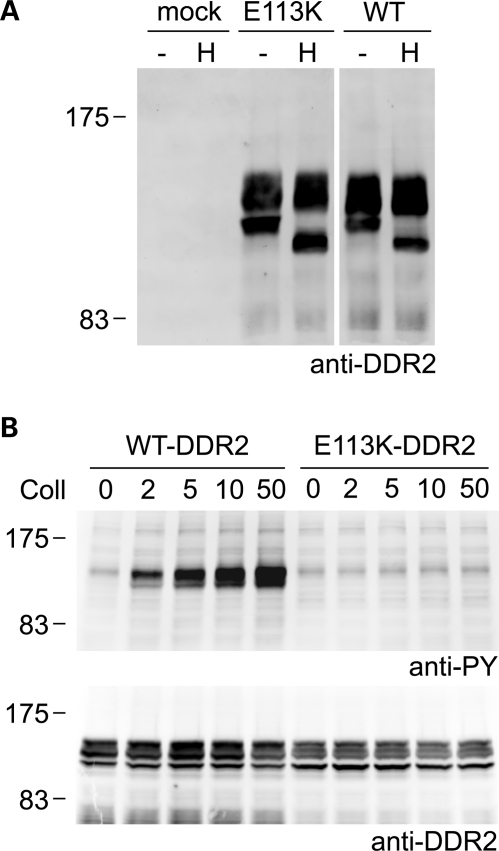
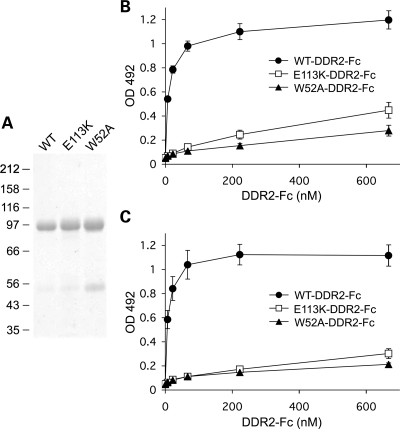
Spondylo-meta-epiphyseal dysplasia (SMED) with short limbs and abnormal calcifications (SMED-SL) is a rare, autosomal recessive human growth disorder, characterized by disproportionate short stature, short limbs, short broad fingers, abnormal metaphyses and epiphyses, platyspondyly and premature calcifications. Recently, three missense mutations and one splice-site mutation in the DDR2 gene were identified as causative genetic defects for SMED-SL, but the underlying cellular and biochemical mechanisms were not explored. Here we report a novel DDR2 missense mutation, c.337G>A (p.E113K), that causes SMED-SL in two siblings in the United Arab Emirates. Another DDR2 missense mutation, c.2254C>T (p.R752C), matching one of the previously reported SMED-SL mutations, was found in a second affected family. DDR2 is a plasma membrane receptor tyrosine kinase that functions as a collagen receptor. We expressed DDR2 constructs with the identified point mutations in human cell lines and evaluated their localization and functional properties. We found that all SMED-SL missense mutants were defective in collagen-induced receptor activation and that the three previously reported mutants (p.T713I, p.I726R and p.R752C) were retained in the endoplasmic reticulum. The novel mutant (p.E113K), in contrast, trafficked normally, like wild-type DDR2, but failed to bind collagen. This finding is in agreement with our recent structural data identifying Glu113 as an important amino acid in the DDR2 ligand-binding site. Our data thus demonstrate that SMED-SL can result from at least two different loss-of-function mechanisms: namely defects in DDR2 targeting to the plasma membrane or the loss of its ligand-binding activity.
短肢-骨干-干骺端发育不良伴短指(趾)和异常钙化(SMED-SL)是一种罕见的常染色体隐性人类生长障碍,其特征为不成比例的身材矮小、短肢、短而宽的手指、骨干和干骺端异常、扁平椎骨和过早钙化。最近,在 DDR2 基因中发现了三个错义突变和一个剪接位点突变,这些突变被认为是 SMED-SL 的致病遗传缺陷,但尚未探讨其潜在的细胞和生化机制。在这里,我们报告了一种新的 DDR2 错义突变,c.337G>A(p.E113K),它导致阿联酋的两个兄弟姐妹患有 SMED-SL。另一个 DDR2 错义突变,c.2254C>T(p.R752C),与之前报道的 SMED-SL 突变之一相匹配,在第二个受影响的家庭中被发现。DDR2 是一种位于质膜上的受体酪氨酸激酶,作为胶原受体发挥作用。我们在人类细胞系中表达了具有鉴定出的点突变的 DDR2 构建体,并评估了它们的定位和功能特性。我们发现,所有 SMED-SL 错义突变体在胶原诱导的受体激活中均存在缺陷,并且之前报道的三个突变体(p.T713I、p.I726R 和 p.R752C)均在内质网中保留。相比之下,新型突变体(p.E113K),像野生型 DDR2 一样正常运输,但不能与胶原结合。这一发现与我们最近的结构数据一致,该数据确定 Glu113 是 DDR2 配体结合位点中的一个重要氨基酸。我们的数据表明,SMED-SL 至少可以由两种不同的功能丧失机制引起:即 DDR2 靶向质膜的缺陷或其配体结合活性的丧失。