Faculty of Computer Science, Dalhousie University, Halifax, Nova Scotia, Canada.
Genome Biol Evol. 2010 Jan 25;2:117-31. doi: 10.1093/gbe/evq004.
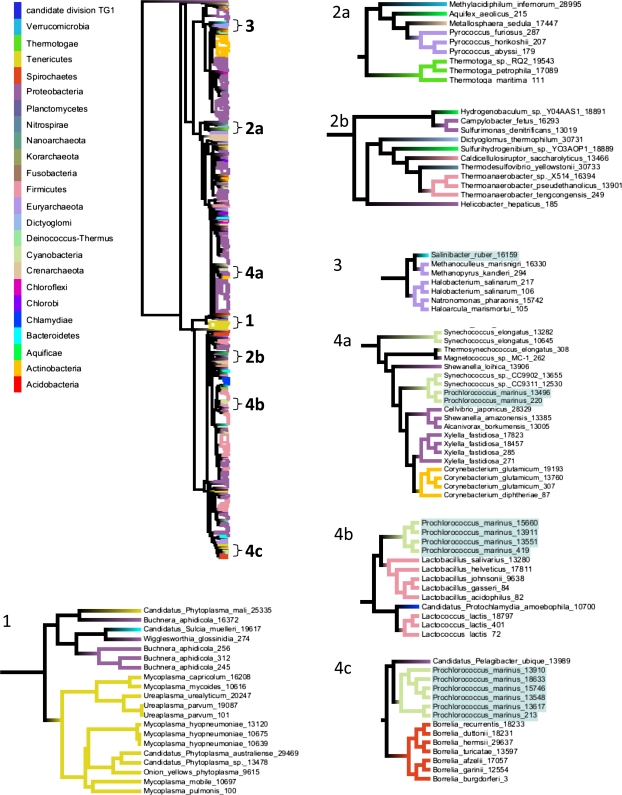
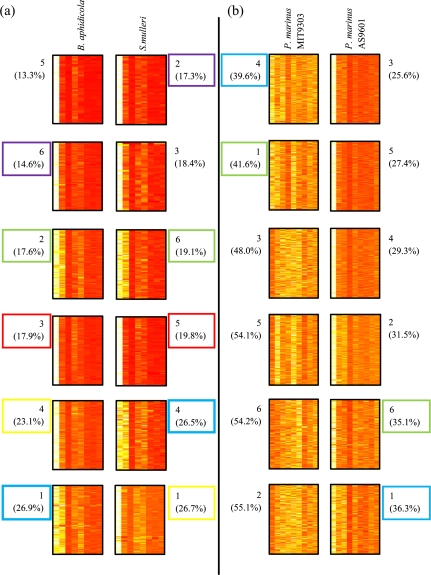
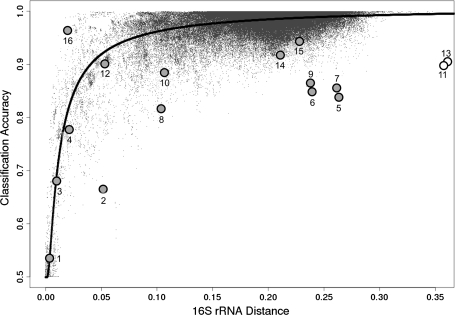
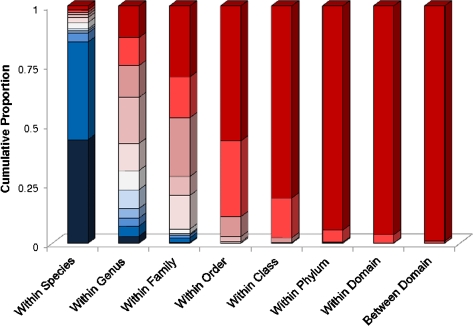
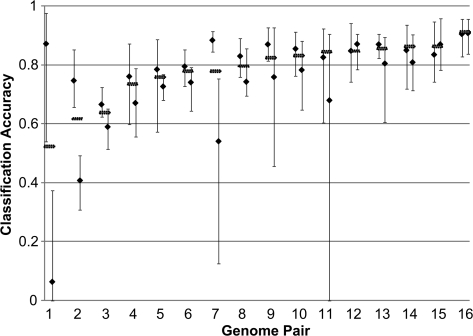
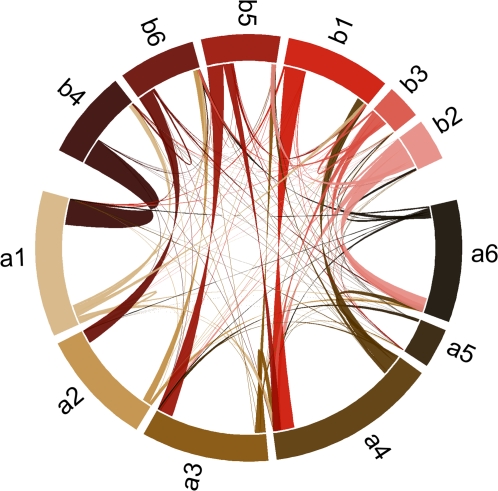
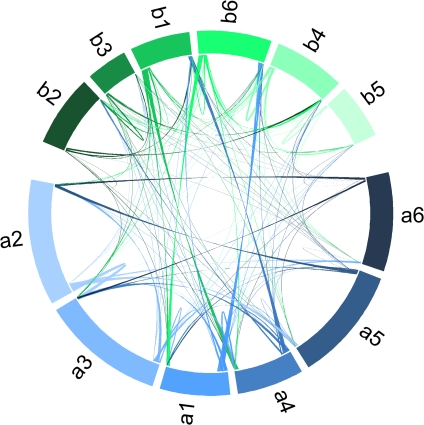
It is well known that patterns of nucleotide composition vary within and among genomes, although the reasons why these variations exist are not completely understood. Between-genome compositional variation has been exploited to assign environmental shotgun sequences to their most likely originating genomes, whereas within-genome variation has been used to identify recently acquired genetic material such as pathogenicity islands. Recent sequence assignment techniques have achieved high levels of accuracy on artificial data sets, but the relative difficulty of distinguishing lineages with varying degrees of relatedness, and different types of genomic sequence, has not been examined in depth. We investigated the compositional differences in a set of 774 sequenced microbial genomes, finding rapid divergence among closely related genomes, but also convergence of compositional patterns among genomes with similar habitats. Support vector machines were then used to distinguish all pairs of genomes based on genome fragments 500 nucleotides in length. The nearly 300,000 accuracy scores obtained from these trials were used to construct general models of distinguishability versus taxonomic and compositional indices of genomic divergence. Unusual genome pairs were evident from their large residuals relative to the fitted model, and we identified several factors including genome reduction, putative lateral genetic transfer, and habitat convergence that influence the distinguishability of genomes. The positional, compositional, and functional context of a fragment within a genome has a strong influence on its likelihood of correct classification, but in a way that depends on the taxonomic and ecological similarity of the comparator genome.
众所周知,核苷酸组成模式在基因组内和基因组之间都存在差异,尽管这些差异存在的原因尚未完全了解。基因组间组成的变化已被用于将环境 shotgun 序列分配给其最可能的起源基因组,而基因组内的变化则被用于识别最近获得的遗传物质,如致病性岛。最近的序列分配技术在人工数据集上达到了很高的准确性水平,但区分具有不同亲缘关系程度和不同类型基因组序列的谱系的相对难度尚未深入研究。我们研究了一组 774 个测序微生物基因组中的组成差异,发现密切相关的基因组之间存在快速分歧,但也发现了具有相似生境的基因组之间组成模式的趋同。然后,支持向量机被用于根据 500 个核苷酸长的基因组片段区分所有基因组对。从这些试验中获得的近 30 万个准确性得分被用于构建可区分性与基因组分歧的分类和组成指标的通用模型。不寻常的基因组对从它们相对于拟合模型的大残差中明显看出,我们确定了几个因素,包括基因组缩减、可能的横向基因转移和生境趋同,这些因素影响基因组的可区分性。基因组内片段的位置、组成和功能上下文对其正确分类的可能性有很强的影响,但影响方式取决于比较基因组的分类和生态相似性。