Diaz Naryttza N, Krause Lutz, Goesmann Alexander, Niehaus Karsten, Nattkemper Tim W
Center for Biotechnology (CeBiTec), Bielefeld University, Bielefeld, Germany.
BMC Bioinformatics. 2009 Feb 11;10:56. doi: 10.1186/1471-2105-10-56.
Metagenomics, or the sequencing and analysis of collective genomes (metagenomes) of microorganisms isolated from an environment, promises direct access to the "unculturable majority". This emerging field offers the potential to lay solid basis on our understanding of the entire living world. However, the taxonomic classification is an essential task in the analysis of metagenomics data sets that it is still far from being solved. We present a novel strategy to predict the taxonomic origin of environmental genomic fragments. The proposed classifier combines the idea of the k-nearest neighbor with strategies from kernel-based learning.
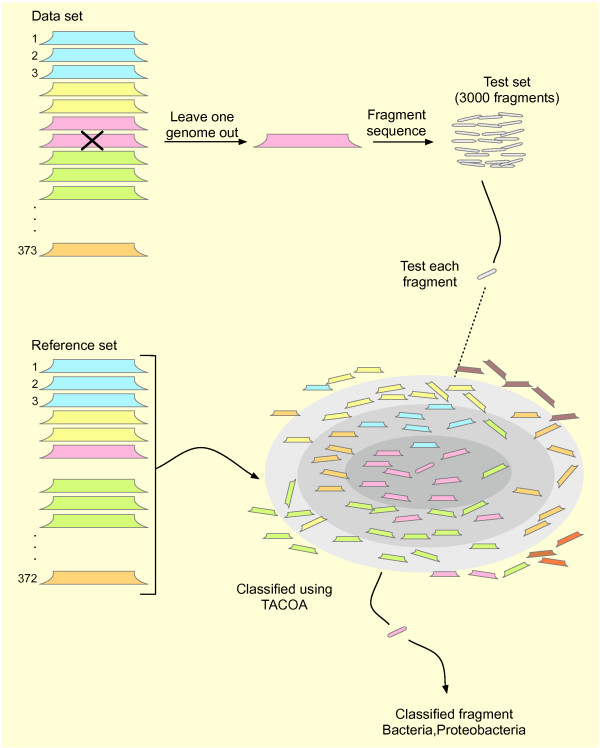
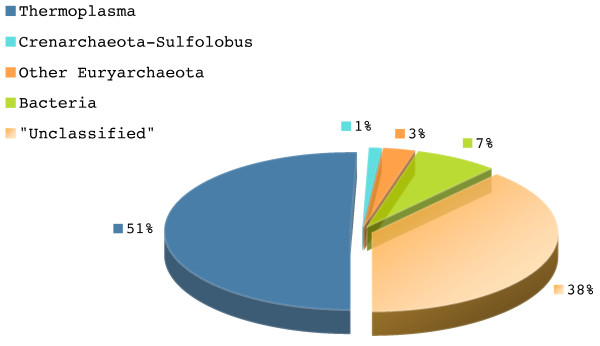
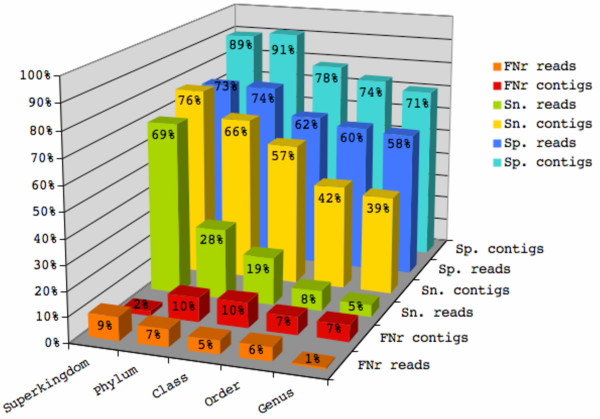
Our novel strategy was extensively evaluated using the leave-one-out cross validation strategy on fragments of variable length (800 bp - 50 Kbp) from 373 completely sequenced genomes. TACOA is able to classify genomic fragments of length 800 bp and 1 Kbp with high accuracy until rank class. For longer fragments > or = 3 Kbp accurate predictions are made at even deeper taxonomic ranks (order and genus). Remarkably, TACOA also produces reliable results when the taxonomic origin of a fragment is not represented in the reference set, thus classifying such fragments to its known broader taxonomic class or simply as "unknown". We compared the classification accuracy of TACOA with the latest intrinsic classifier PhyloPythia using 63 recently published complete genomes. For fragments of length 800 bp and 1 Kbp the overall accuracy of TACOA is higher than that obtained by PhyloPythia at all taxonomic ranks. For all fragment lengths, both methods achieved comparable high specificity results up to rank class and low false negative rates are also obtained.
An accurate multi-class taxonomic classifier was developed for environmental genomic fragments. TACOA can predict with high reliability the taxonomic origin of genomic fragments as short as 800 bp. The proposed method is transparent, fast, accurate and the reference set can be easily updated as newly sequenced genomes become available. Moreover, the method demonstrated to be competitive when compared to the most current classifier PhyloPythia and has the advantage that it can be locally installed and the reference set can be kept up-to-date.
宏基因组学,即对从环境中分离出的微生物的集体基因组(宏基因组)进行测序和分析,有望直接接触到“绝大多数不可培养的微生物”。这个新兴领域为我们理解整个生物世界奠定坚实基础提供了潜力。然而,分类学分类是宏基因组学数据集分析中的一项基本任务,目前仍远未得到解决。我们提出了一种预测环境基因组片段分类学来源的新策略。所提出的分类器将k近邻算法的思想与基于核学习的策略相结合。
我们使用留一法交叉验证策略,对来自373个完全测序基因组的可变长度(800bp - 50Kbp)片段进行了广泛评估。TACOA能够以高精度将长度为800bp和1Kbp的基因组片段分类到等级分类。对于长度大于或等于3Kbp的较长片段,在更深的分类学等级(目和属)上也能做出准确预测。值得注意的是,当片段的分类学来源在参考集中未被代表时,TACOA也能产生可靠的结果,从而将此类片段分类到其已知的更宽泛的分类类别或简单地归类为“未知”。我们使用63个最近发表的完整基因组,将TACOA的分类准确性与最新的内在分类器PhyloPythia进行了比较。对于长度为800bp和1Kbp的片段,TACOA在所有分类学等级上的总体准确性都高于PhyloPythia。对于所有片段长度,两种方法在等级分类之前都获得了相当高的特异性结果,并且假阴性率也很低。
开发了一种用于环境基因组片段的准确多类分类器。TACOA能够以高可靠性预测短至800bp的基因组片段的分类学来源。所提出的方法透明、快速、准确,并且随着新测序基因组的出现,参考集可以很容易地更新。此外,与当前最先进的分类器PhyloPythia相比,该方法具有竞争力,并且具有可以在本地安装且参考集可以保持最新的优势。