The Institute of Mathematical Sciences, Chennai, Tamil Nadu, India.
PLoS One. 2010 Mar 22;5(3):e9722. doi: 10.1371/journal.pone.0009722.
Identifying transcription factor binding sites (TFBS) in silico is key in understanding gene regulation. TFBS are string patterns that exhibit some variability, commonly modelled as "position weight matrices" (PWMs). Though convenient, the PWM has significant limitations, in particular the assumed independence of positions within the binding motif; and predictions based on PWMs are usually not very specific to known functional sites. Analysis here on binding sites in yeast suggests that correlation of dinucleotides is not limited to near-neighbours, but can extend over considerable gaps.
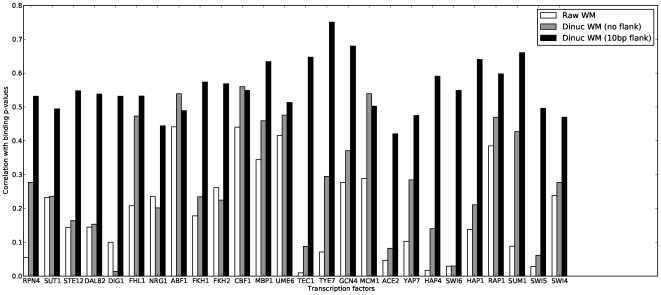
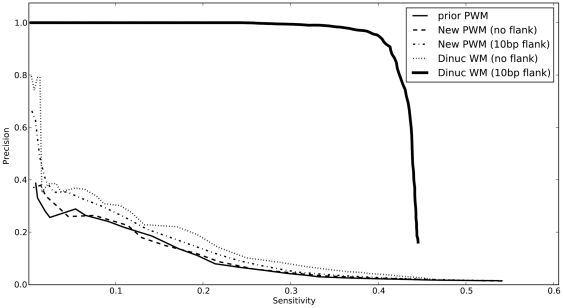
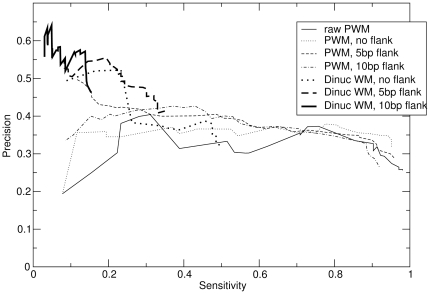
METHODOLOGY/PRINCIPAL FINDINGS: I describe a straightforward generalization of the PWM model, that considers frequencies of dinucleotides instead of individual nucleotides. Unlike previous efforts, this method considers all dinucleotides within an extended binding region, and does not make an attempt to determine a priori the significance of particular dinucleotide correlations. I describe how to use a "dinucleotide weight matrix" (DWM) to predict binding sites, dealing in particular with the complication that its entries are not independent probabilities. Benchmarks show, for many factors, a dramatic improvement over PWMs in precision of predicting known targets. In most cases, significant further improvement arises by extending the commonly defined "core motifs" by about 10 bp on either side. Though this flanking sequence shows no strong motif at the nucleotide level, the predictive power of the dinucleotide model suggests that the "signature" in DNA sequence of protein-binding affinity extends beyond the core protein-DNA contact region.
CONCLUSION/SIGNIFICANCE: While computationally more demanding and slower than PWM-based approaches, this dinucleotide method is straightforward, both conceptually and in implementation, and can serve as a basis for future improvements.
在计算机中识别转录因子结合位点(TFBS)是理解基因调控的关键。TFBS 是表现出一定可变性的字符串模式,通常被建模为“位置权重矩阵”(PWMs)。尽管 PWMs 很方便,但它有很大的局限性,特别是绑定基序内位置的假设独立性;并且基于 PWMs 的预测通常对已知功能位点不是很具体。对酵母中结合位点的分析表明,二核苷酸的相关性不仅限于近邻,而是可以延伸到相当大的间隙。
方法/主要发现:我描述了 PWM 模型的一种直接推广,该模型考虑了二核苷酸的频率而不是单个核苷酸。与以前的努力不同,这种方法考虑了扩展的绑定区域内的所有二核苷酸,并且不尝试预先确定特定二核苷酸相关性的重要性。我描述了如何使用“二核苷酸权重矩阵”(DWM)来预测结合位点,特别是处理其条目不是独立概率的复杂性。基准测试表明,对于许多因素,与 PWM 相比,在预测已知目标的精度方面有了显著提高。在大多数情况下,通过在通常定义的“核心基序”的任一侧扩展约 10 bp,可以进一步显著提高。尽管这种侧翼序列在核苷酸水平上没有强烈的基序,但二核苷酸模型的预测能力表明,蛋白质结合亲和力在 DNA 序列中的“特征”超出了核心蛋白-DNA 接触区域。
结论/意义:虽然这种二核苷酸方法在计算上比基于 PWM 的方法要求更高且更慢,但它在概念上和实现上都很简单,可以作为未来改进的基础。