Siddharthan Rahul
The Institute of Mathematical Sciences, Chennai, India.
PLoS Comput Biol. 2008 Aug 29;4(8):e1000156. doi: 10.1371/journal.pcbi.1000156.
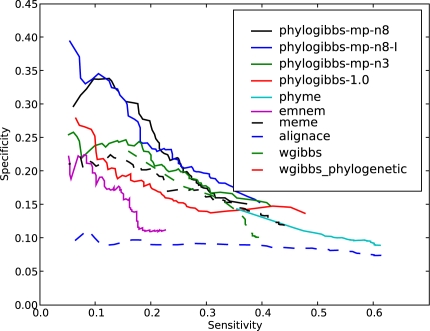
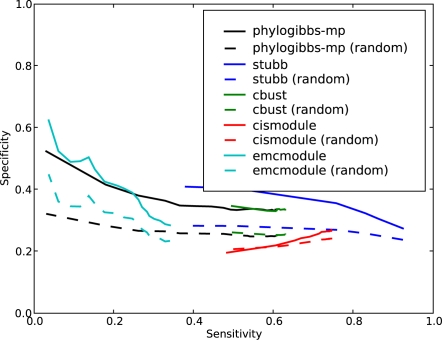
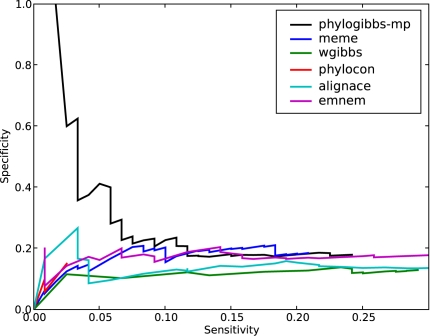
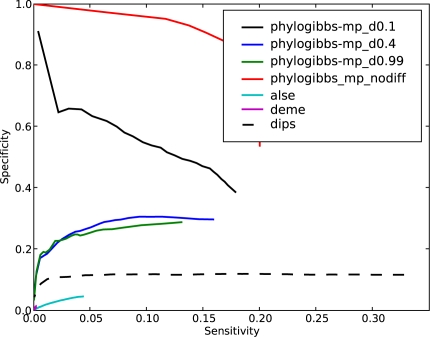
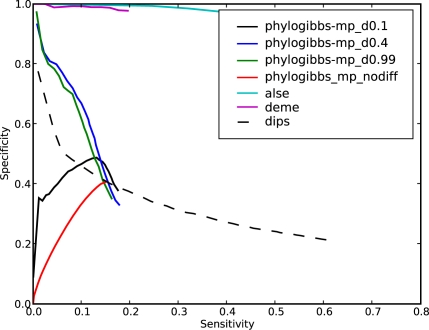
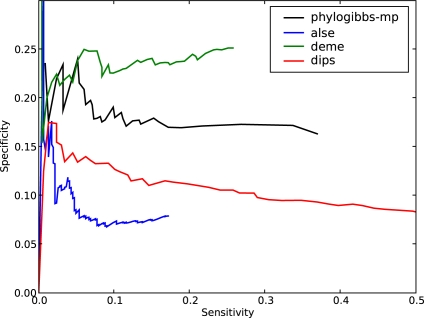
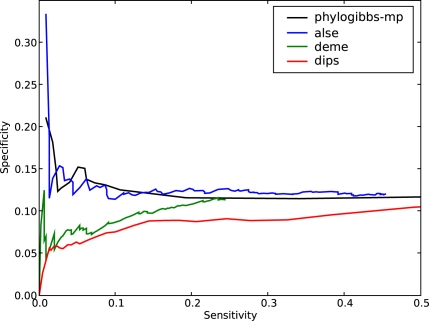
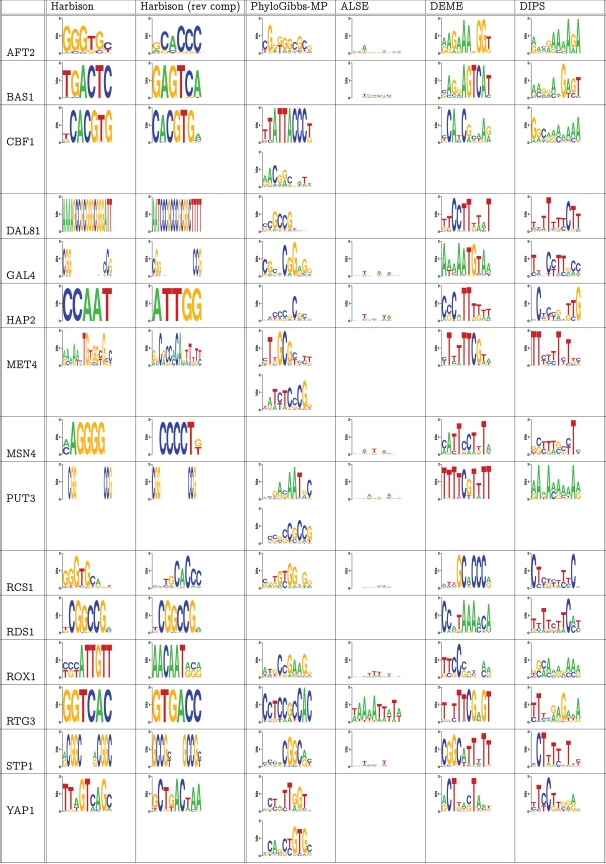
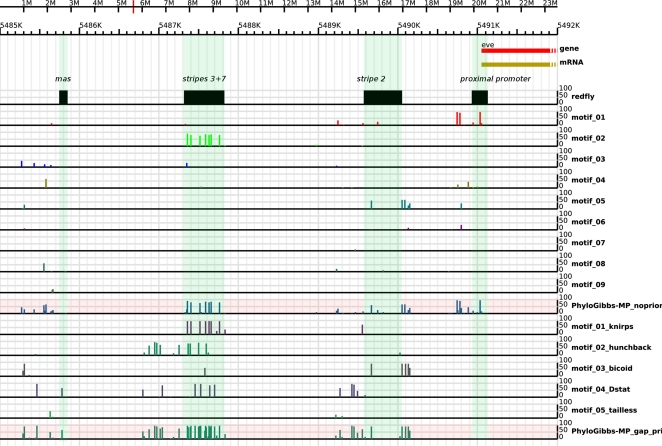
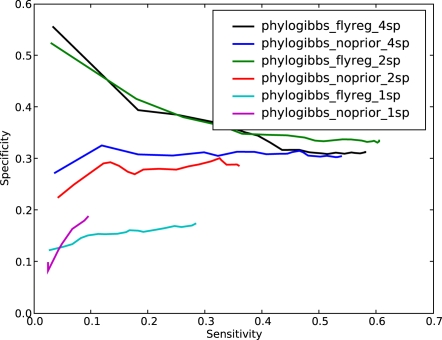
PhyloGibbs, our recent Gibbs-sampling motif-finder, takes phylogeny into account in detecting binding sites for transcription factors in DNA and assigns posterior probabilities to its predictions obtained by sampling the entire configuration space. Here, in an extension called PhyloGibbs-MP, we widen the scope of the program, addressing two major problems in computational regulatory genomics. First, PhyloGibbs-MP can localise predictions to small, undetermined regions of a large input sequence, thus effectively predicting cis-regulatory modules (CRMs) ab initio while simultaneously predicting binding sites in those modules-tasks that are usually done by two separate programs. PhyloGibbs-MP's performance at such ab initio CRM prediction is comparable with or superior to dedicated module-prediction software that use prior knowledge of previously characterised transcription factors. Second, PhyloGibbs-MP can predict motifs that differentiate between two (or more) different groups of regulatory regions, that is, motifs that occur preferentially in one group over the others. While other "discriminative motif-finders" have been published in the literature, PhyloGibbs-MP's implementation has some unique features and flexibility. Benchmarks on synthetic and actual genomic data show that this algorithm is successful at enhancing predictions of differentiating sites and suppressing predictions of common sites and compares with or outperforms other discriminative motif-finders on actual genomic data. Additional enhancements include significant performance and speed improvements, the ability to use "informative priors" on known transcription factors, and the ability to output annotations in a format that can be visualised with the Generic Genome Browser. In stand-alone motif-finding, PhyloGibbs-MP remains competitive, outperforming PhyloGibbs-1.0 and other programs on benchmark data.
我们最新的吉布斯采样基序查找工具PhyloGibbs在检测DNA中转录因子的结合位点时会考虑系统发育,并为通过对整个配置空间进行采样获得的预测赋予后验概率。在此,在一个名为PhyloGibbs-MP的扩展版本中,我们拓宽了该程序的范围,解决了计算调控基因组学中的两个主要问题。首先,PhyloGibbs-MP可以将预测定位到大型输入序列的小的未确定区域,从而有效地从头预测顺式调控模块(CRM),同时预测这些模块中的结合位点——这些任务通常由两个单独的程序完成。PhyloGibbs-MP在这种从头CRM预测方面的性能与使用先前表征的转录因子的先验知识的专用模块预测软件相当或更优。其次,PhyloGibbs-MP可以预测区分两个(或更多)不同调控区域组的基序,即优先在一组中而非其他组中出现的基序。虽然文献中已经发表了其他“判别性基序查找工具”,但PhyloGibbs-MP的实现具有一些独特的特性和灵活性。对合成和实际基因组数据的基准测试表明,该算法在增强区分位点的预测和抑制常见位点的预测方面是成功的,并且在实际基因组数据上与其他判别性基序查找工具相当或更优。其他改进包括显著的性能和速度提升、对已知转录因子使用“信息先验”的能力以及以可通过通用基因组浏览器可视化的格式输出注释的能力。在独立的基序查找中,PhyloGibbs-MP仍然具有竞争力,在基准数据上优于PhyloGibbs-1.0和其他程序。