School of Biological Sciences, University of Nebraska - Lincoln, NE 68588-0118, USA.
BMC Evol Biol. 2010 Mar 31;10:90. doi: 10.1186/1471-2148-10-90.
Exon-primed intron-crossing (EPIC) markers have three advantages over anonymous genomic sequences in studying evolution of natural populations. First, the universal primers designed in exon regions can be applied across a broad taxonomic range. Second, the homology of EPIC-amplified sequences can be easily determined by comparing either their exon or intron portion depending on the genetic distance between the taxa. Third, having both the exon and intron fragments could help in examining genetic variation at the intraspecific and interspecific level simultaneously, particularly helpful when studying species complex. However, the paucity of EPIC markers has hindered multilocus studies using nuclear gene sequences, particularly in teleost fishes.
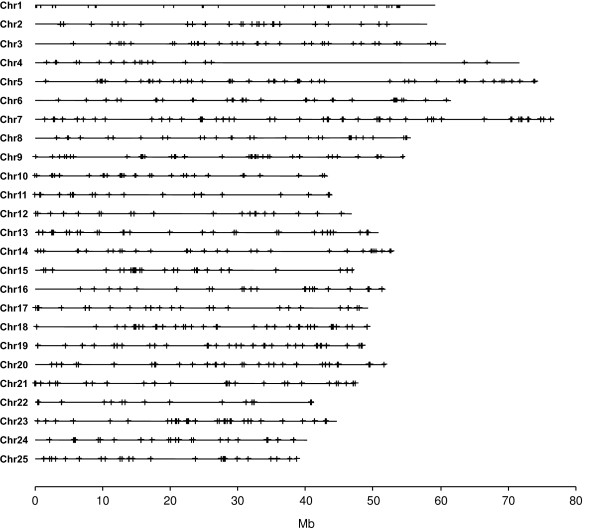
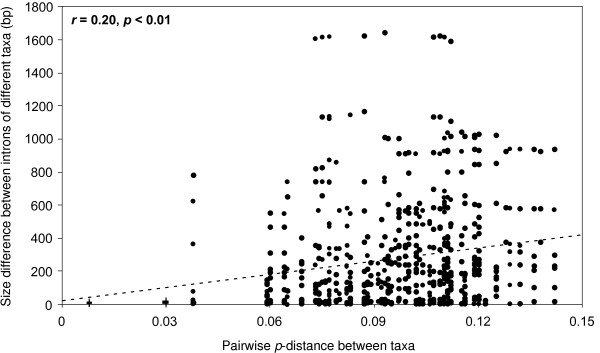
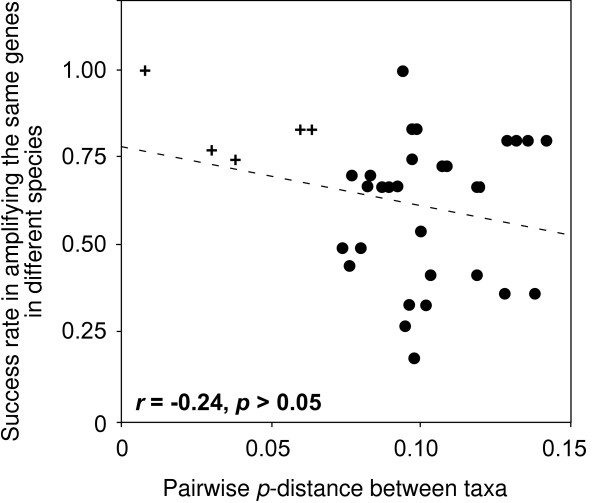
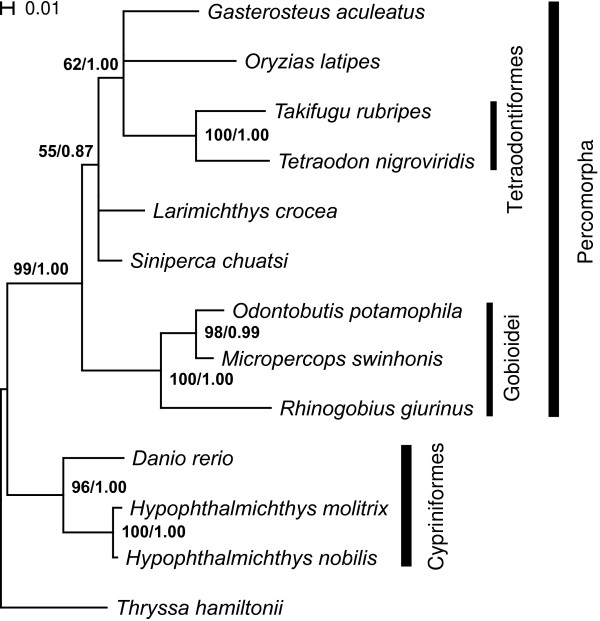
We introduce a bioinformatics pipeline for developing EPIC markers by comparing the whole genome sequences between two or more species. By applying this approach on five teleost fishes whose genomes were available in the Ensembl database http://www.ensembl.org, we identified 210 EPIC markers that have single-copy and conserved exon regions with identity greater than 85% among the five teleost fishes. We tested 12 randomly chosen EPIC markers in nine teleost species having a wide phylogenetic range. The success rate of amplifying and sequencing those markers varied from 44% to 100% in different species. We analyzed the exon sequences of the 12 EPIC markers from 13 teleosts. The resulting phylogeny contains many traditionally well-supported clades, indicating the usefulness of the exon portion of EPIC markers in reconstructing species phylogeny, in addition to the value of the intron portion of EPIC markers in interrogating the population history.
This study illustrated an effective approach to develop EPIC markers in a taxonomic group, where two or more genome sequences are available. The markers identified could be amplified across a broad taxonomic range of teleost fishes. The phylogenetic utility of individual markers varied according to intron size and amplifiability. The bioinformatics pipelines developed are readily adapted to other taxonomic groups.
在研究自然种群的进化时,exon-primed intron-crossing(EPIC)标记相对于匿名基因组序列有三个优势。首先,在exon 区域设计的通用引物可以应用于广泛的分类范围内。其次,根据分类群之间的遗传距离,通过比较 EPIC 扩增序列的exon 或 intron 部分,很容易确定其同源性。第三,拥有exon 和 intron 片段可以帮助同时检查种内和种间水平的遗传变异,在研究物种复合体时特别有用。然而,EPIC 标记的缺乏阻碍了使用核基因序列进行多位点研究,特别是在硬骨鱼类中。
我们通过比较两个或更多物种的全基因组序列,引入了一种用于开发 EPIC 标记的生物信息学管道。通过将这种方法应用于五个基因组可在 Ensembl 数据库(http://www.ensembl.org)中获得的硬骨鱼类,我们鉴定了 210 个具有单一拷贝和保守 exon 区域的 EPIC 标记,这些标记在这五种硬骨鱼类之间的同一性大于 85%。我们在具有广泛系统发育范围的 9 个硬骨鱼类中测试了 12 个随机选择的 EPIC 标记。在不同的物种中,这些标记的扩增和测序成功率从 44%到 100%不等。我们分析了 13 个硬骨鱼类中 12 个 EPIC 标记的 exon 序列。得到的系统发育树包含许多传统上支持良好的分支,表明 EPIC 标记的 exon 部分在重建物种系统发育方面有用,除了 EPIC 标记的 intron 部分在探究种群历史方面的价值。
本研究说明了在有两个或更多基因组序列可用的分类群中开发 EPIC 标记的有效方法。鉴定出的标记可以在广泛的硬骨鱼类分类范围内扩增。单个标记的系统发育用途根据 intron 大小和可扩增性而有所不同。开发的生物信息学管道可以很容易地适应其他分类群。