Costa Igor Rodrigues, Prosdocimi Francisco, Jennings W Bryan
Laboratório de Genômica e Biodiversidade, Instituto de Bioquímica Médica Leopoldo de Meis, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ, 21941-902, Brazil;
Departamento de Vertebrados, Museu Nacional, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ, 20940-040, Brazil.
Genome Res. 2016 Sep;26(9):1257-67. doi: 10.1101/gr.203950.115. Epub 2016 Jul 19.
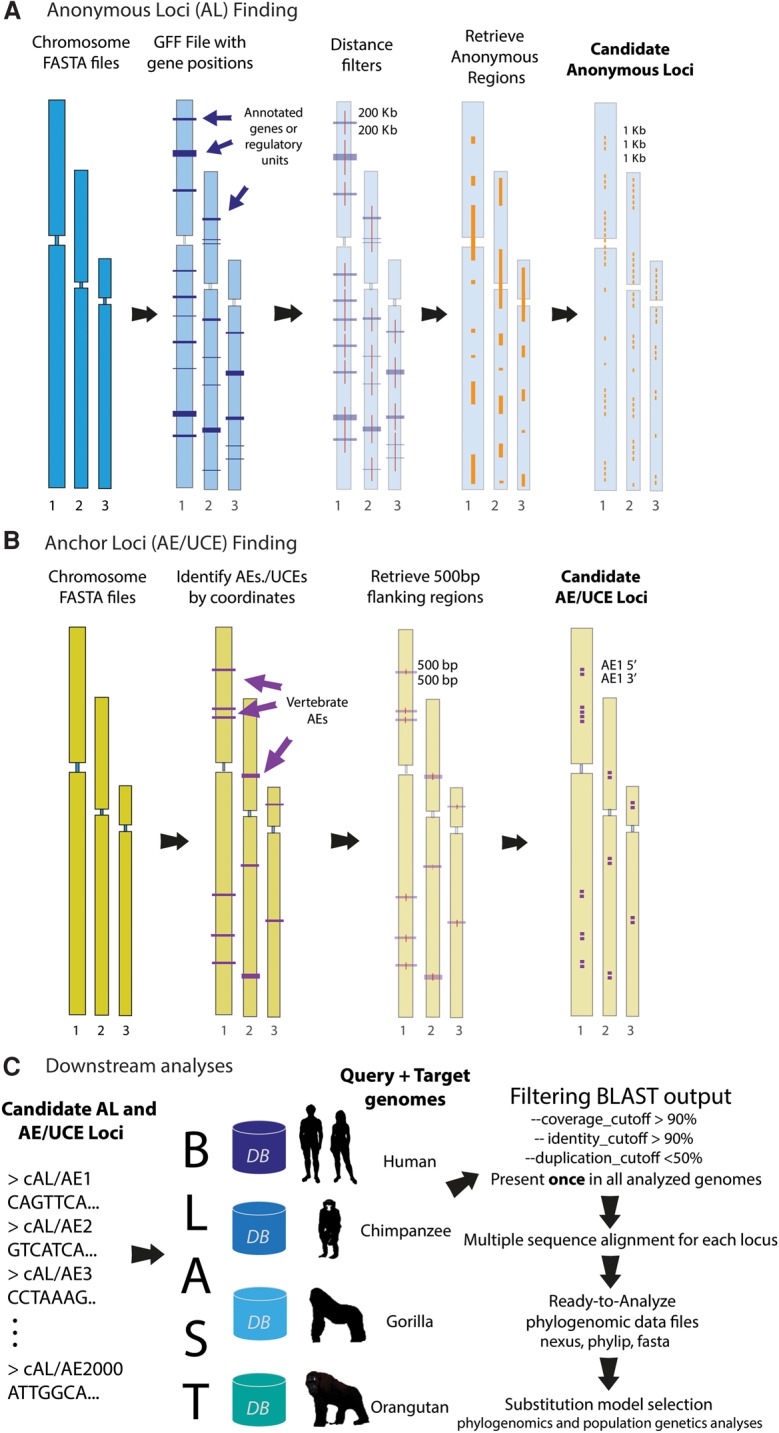
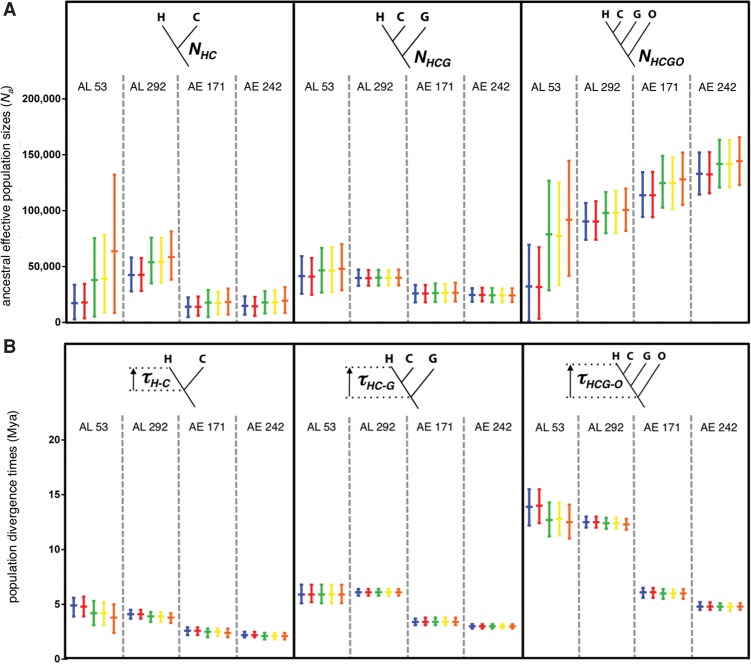
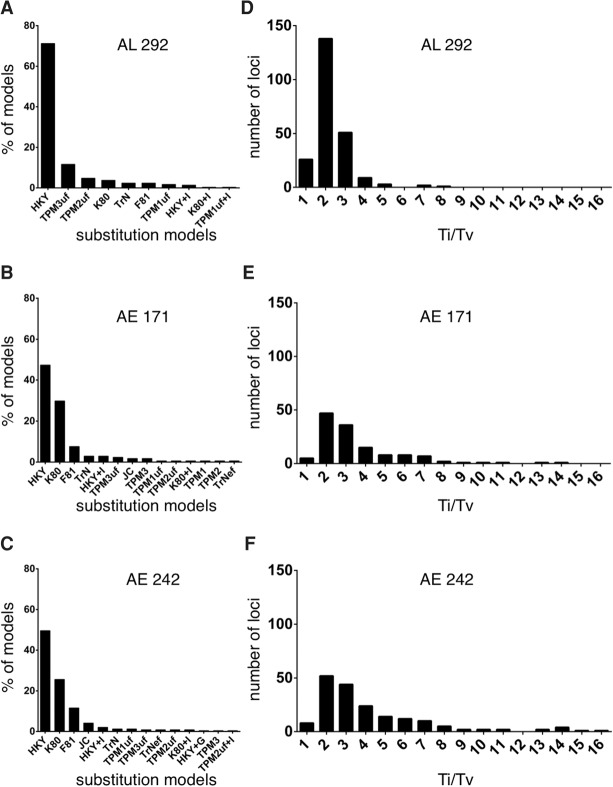
The increasing availability of complete genome data is facilitating the acquisition of phylogenomic data sets, but the process of obtaining orthologous sequences from other genomes and assembling multiple sequence alignments remains piecemeal and arduous. We designed software that performs these tasks and outputs anonymous loci (AL) or anchored enrichment/ultraconserved element loci (AE/UCE) data sets in ready-to-analyze formats. We demonstrate our program by applying it to the hominoids. Starting with human, chimpanzee, gorilla, and orangutan genomes, our software generated an exhaustive data set of 292 ALs (∼1 kb each) in ∼3 h. Not only did analyses of our AL data set validate the program by yielding a portrait of hominoid evolution in agreement with previous studies, but the accuracy and precision of our estimated ancestral effective population sizes and speciation times represent improvements. We also used our program with a published set of 512 vertebrate-wide AE "probe" sequences to generate data sets consisting of 171 and 242 independent loci (∼1 kb each) in 11 and 13 min, respectively. The former data set consisted of flanking sequences 500 bp from adjacent AEs, while the latter contained sequences bordering AEs. Although our AE data sets produced the expected hominoid species tree, coalescent-based estimates of ancestral population sizes and speciation times based on these data were considerably lower than estimates from our AL data set and previous studies. Accordingly, we suggest that loci subjected to direct or indirect selection may not be appropriate for coalescent-based methods. Complete in silico approaches, combined with the burgeoning genome databases, will accelerate the pace of phylogenomics.
全基因组数据日益容易获取,这推动了系统发育基因组数据集的获取,但从其他基因组中获取直系同源序列并组装多序列比对的过程仍然是零碎且艰巨的。我们设计了一款软件,该软件能执行这些任务,并以易于分析的格式输出匿名基因座(AL)或锚定富集/超保守元件基因座(AE/UCE)数据集。我们通过将该程序应用于类人猿来展示我们的程序。从人类、黑猩猩、大猩猩和猩猩的基因组开始,我们的软件在约3小时内生成了一个包含292个AL(每个约1 kb)的详尽数据集。对我们的AL数据集的分析不仅通过得出与先前研究一致的类人猿进化图景验证了该程序,而且我们估计的祖先有效种群大小和物种形成时间的准确性和精确性也有所提高。我们还将我们的程序与一组已发表的512个全脊椎动物AE“探针”序列一起使用,分别在11分钟和13分钟内生成了由171个和242个独立基因座(每个约1 kb)组成的数据集。前一个数据集由来自相邻AE的500 bp侧翼序列组成,而后一个数据集包含与AE相邻的序列。尽管我们的AE数据集产生了预期的类人猿物种树,但基于这些数据的基于溯祖法的祖先种群大小和物种形成时间估计值明显低于我们的AL数据集和先前研究的估计值。因此,我们认为受到直接或间接选择的基因座可能不适用于基于溯祖法的方法。完整的计算机方法,结合迅速发展的基因组数据库,将加快系统发育基因组学的发展步伐。