Genetics Unit, de Ciències Experimentals i de la Salut, Universitat Pompeu Fabra, Barcelona, Spain.
BMC Med Genet. 2010 Apr 19;11:61. doi: 10.1186/1471-2350-11-61.
GTF2I codes for a general intrinsic transcription factor and calcium channel regulator TFII-I, with high and ubiquitous expression, and a strong candidate for involvement in the morphological and neuro-developmental anomalies of the Williams-Beuren syndrome (WBS). WBS is a genetic disorder due to a recurring deletion of about 1,55-1,83 Mb containing 25-28 genes in chromosome band 7q11.23 including GTF2I. Completed homozygous loss of either the Gtf2i or Gtf2ird1 function in mice provided additional evidence for the involvement of both genes in the craniofacial and cognitive phenotype. Unfortunately nothing is now about the behavioral characterization of heterozygous mice.
By gene targeting we have generated a mutant mice with a deletion of the first 140 amino-acids of TFII-I. mRNA and protein expression analysis were used to document the effect of the study deletion. We performed behavioral characterization of heterozygous mutant mice to document in vivo implications of TFII-I in the cognitive profile of WBS patients.
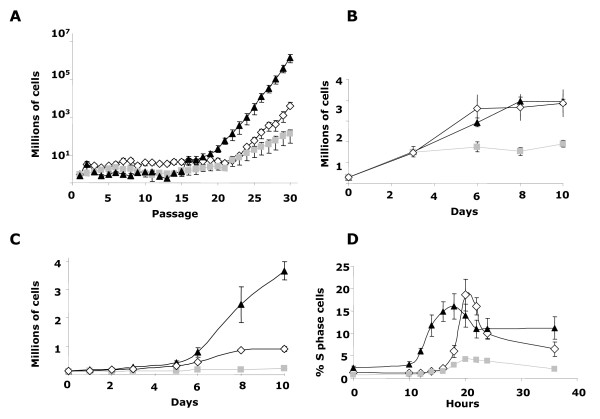
Homozygous and heterozygous mutant mice exhibit craniofacial alterations, most clearly represented in homozygous condition. Behavioral test demonstrate that heterozygous mutant mice exhibit some neurobehavioral alterations and hyperacusis or odynacusis that could be associated with specific features of WBS phenotype. Homozygous mutant mice present highly compromised embryonic viability and fertility. Regarding cellular model, we documented a retarded growth in heterozygous MEFs respect to homozygous or wild-type MEFs.
Our data confirm that, although additive effects of haploinsufficiency at several genes may contribute to the full craniofacial or neurocognitive features of WBS, correct expression of GTF2I is one of the main players. In addition, these findings show that the deletion of the fist 140 amino-acids of TFII-I altered it correct function leading to a clear phenotype, at both levels, at the cellular model and at the in vivo model.
GTF2I 编码一种普遍存在的内在转录因子和钙通道调节因子 TFII-I,具有高表达和广泛表达,是参与威廉姆斯-比伦综合征(WBS)形态和神经发育异常的强有力候选基因。WBS 是一种遗传性疾病,由于大约 1,55-1,83 Mb 的重复缺失,导致染色体 7q11.23 带上的 25-28 个基因缺失,包括 GTF2I。在小鼠中完成 Gtf2i 或 Gtf2ird1 功能的纯合缺失提供了更多证据表明这两个基因都参与了颅面和认知表型。不幸的是,现在还没有关于杂合子小鼠行为特征的研究。
通过基因靶向,我们生成了一种缺失 TFII-I 前 140 个氨基酸的突变小鼠。使用 mRNA 和蛋白质表达分析来记录研究缺失的影响。我们对杂合突变小鼠进行了行为特征分析,以记录 TFII-I 在 WBS 患者认知特征中的体内影响。
纯合和杂合突变小鼠均表现出颅面改变,在纯合条件下最为明显。行为测试表明,杂合突变小鼠表现出一些神经行为改变和听觉过敏或味觉过敏,这可能与 WBS 表型的特定特征有关。纯合突变小鼠的胚胎活力和生育力受到严重损害。关于细胞模型,我们记录到杂合 MEFs 的生长速度比纯合或野生型 MEFs 慢。
我们的数据证实,尽管几个基因的杂合子缺失的加性效应可能有助于 WBS 的全部颅面或神经认知特征,但 GTF2I 的正确表达是主要因素之一。此外,这些发现表明,TFII-I 的前 140 个氨基酸的缺失改变了其正确功能,导致了细胞模型和体内模型中明显的表型。