Interdisciplinary Centre for Bioinformatics, University Leipzig, Germany.
BMC Bioinformatics. 2010 Apr 27;11:207. doi: 10.1186/1471-2105-11-207.
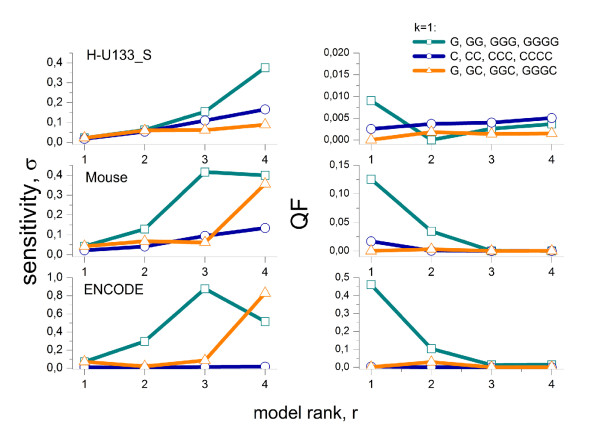
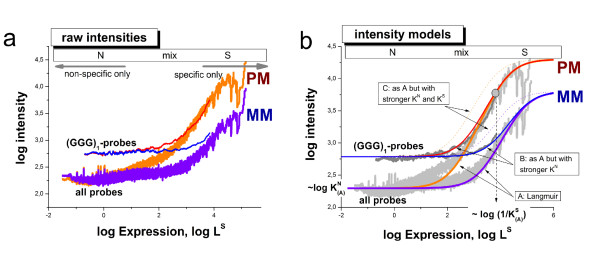
The brightness of the probe spots on expression microarrays intends to measure the abundance of specific mRNA targets. Probes with runs of at least three guanines (G) in their sequence show abnormal high intensities which reflect rather probe effects than target concentrations. This G-bias requires correction prior to downstream expression analysis.
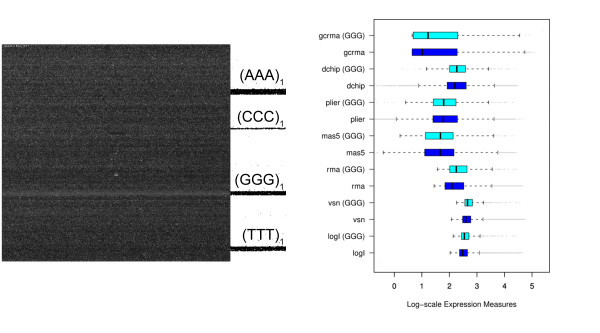
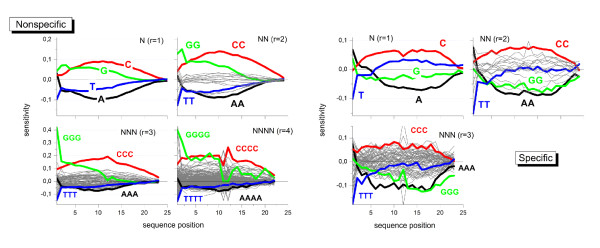
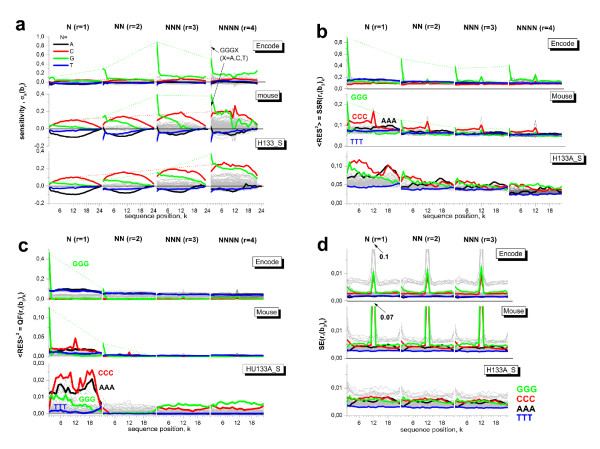
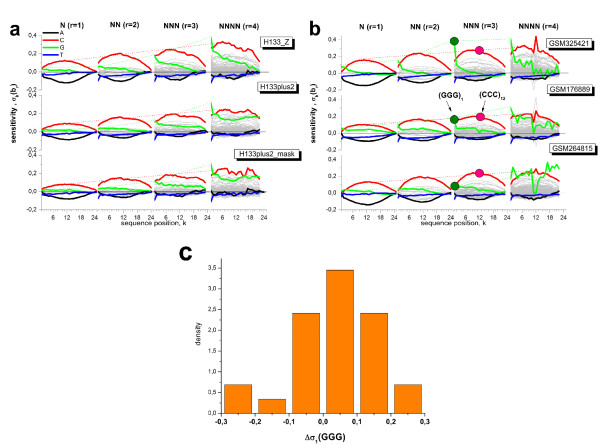
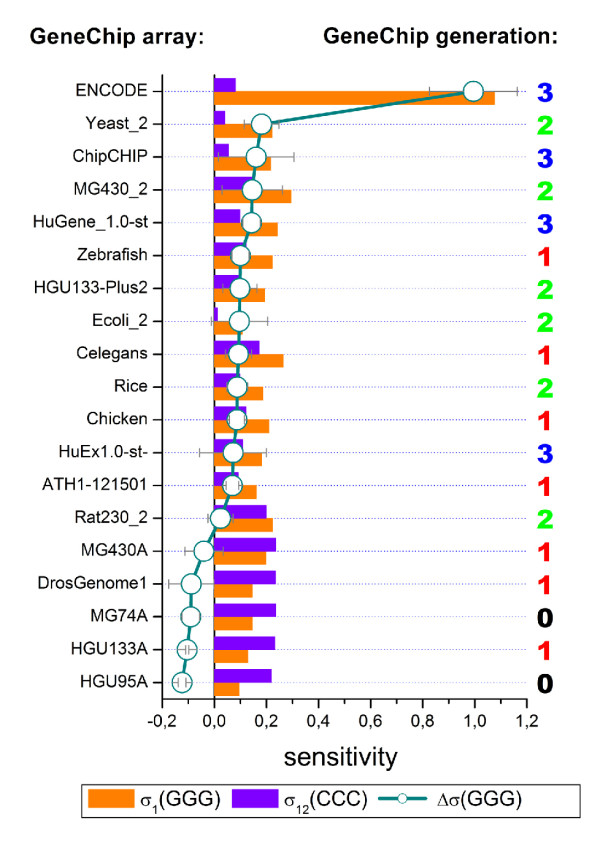
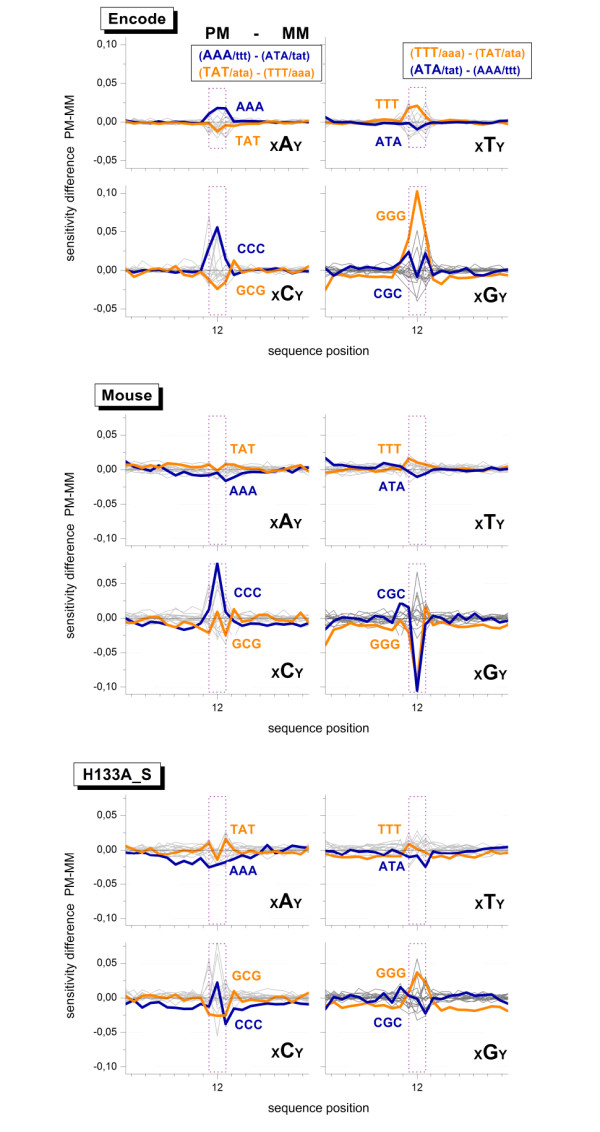
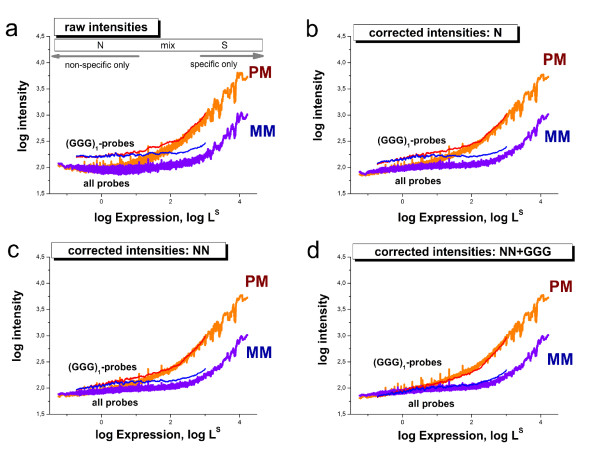
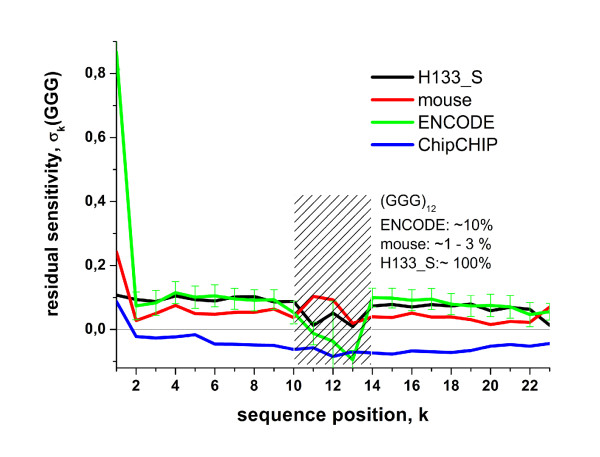
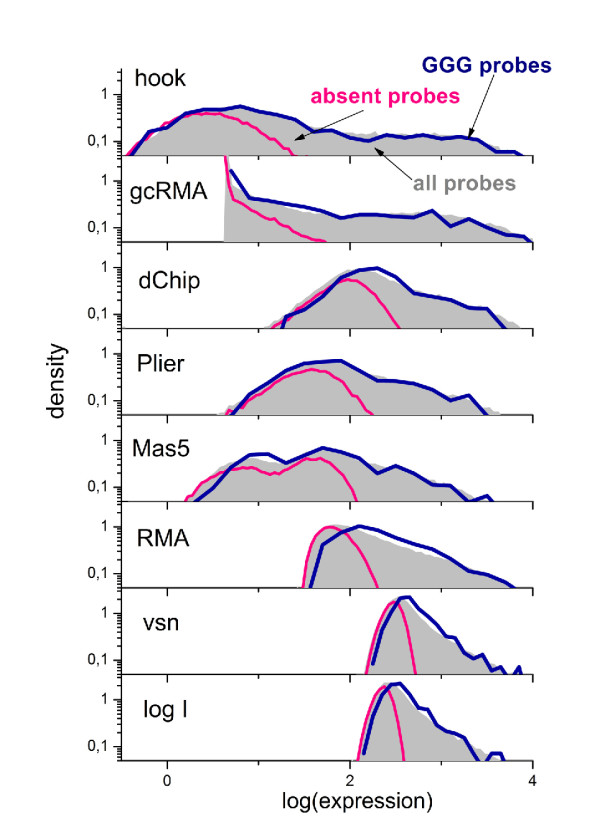
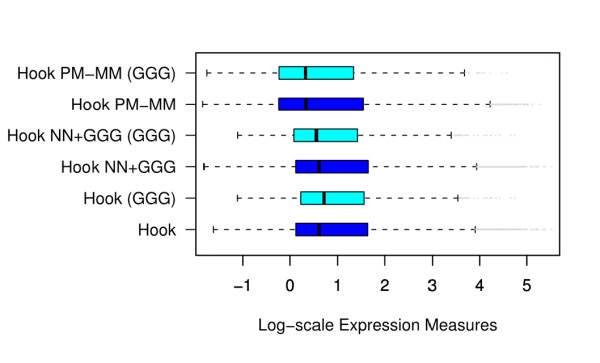
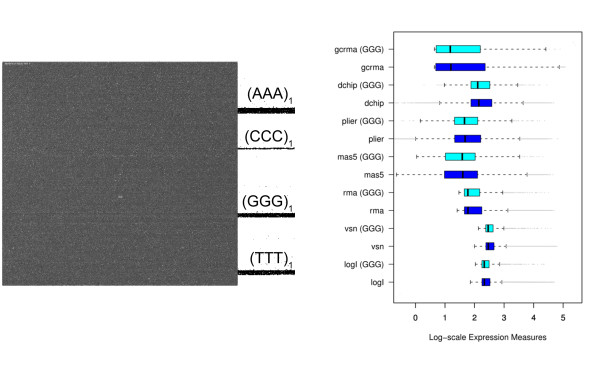
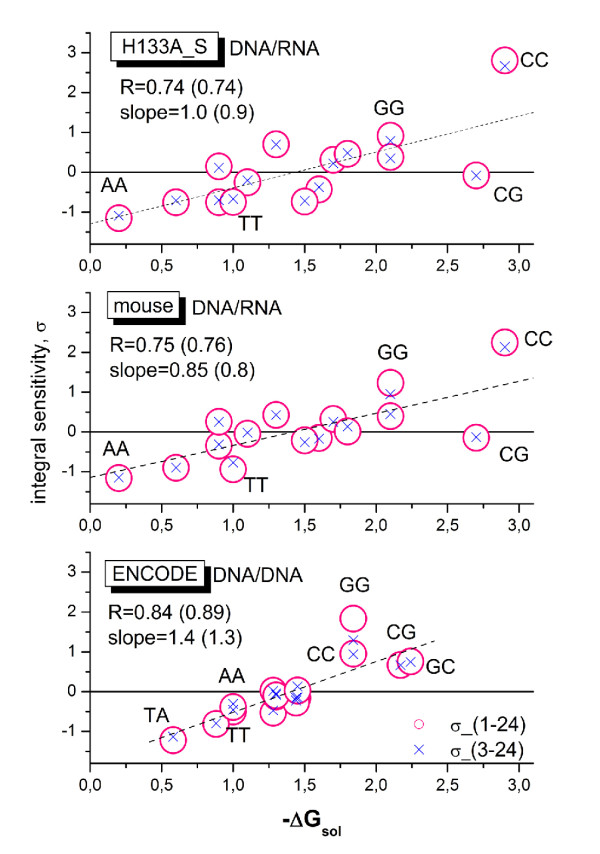
Longer runs of three or more consecutive G along the probe sequence and in particular triple degenerated G at its solution end ((GGG)1-effect) are associated with exceptionally large probe intensities on GeneChip expression arrays. This intensity bias is related to non-specific hybridization and affects both perfect match and mismatch probes. The (GGG)1-effect tends to increase gradually for microarrays of later GeneChip generations. It was found for DNA/RNA as well as for DNA/DNA probe/target-hybridization chemistries. Amplification of sample RNA using T7-primers is associated with strong positive amplitudes of the G-bias whereas alternative amplification protocols using random primers give rise to much smaller and partly even negative amplitudes. We applied positional dependent sensitivity models to analyze the specifics of probe intensities in the context of all possible short sequence motifs of one to four adjacent nucleotides along the 25meric probe sequence. Most of the longer motifs are adequately described using a nearest-neighbor (NN) model. In contrast, runs of degenerated guanines require explicit consideration of next nearest neighbors (GGG terms). Preprocessing methods such as vsn, RMA, dChip, MAS5 and gcRMA only insufficiently remove the G-bias from data.
Positional and motif dependent sensitivity models accounts for sequence effects of oligonucleotide probe intensities. We propose a positional dependent NN+GGG hybrid model to correct the intensity bias associated with probes containing poly-G motifs. It is implemented as a single-chip based calibration algorithm for GeneChips which can be applied in a pre-correction step prior to standard preprocessing.
表达微阵列上探针点的亮度旨在测量特定 mRNA 靶标物的丰度。在其序列中具有至少三个连续鸟嘌呤 (G) 的探针显示出异常高的强度,这反映的与其说是靶浓度,不如说是探针效应。在进行下游表达分析之前,需要对这种 G 偏倚进行校正。
在探针序列中连续三个或更多个连续 G 的较长序列,特别是在其溶液端的三重简并 G((GGG)1-效应)与基因芯片表达阵列上异常大的探针强度相关。这种强度偏差与非特异性杂交有关,影响完全匹配和错配探针。(GGG)1-效应倾向于随着基因芯片代际的推移而逐渐增加。它在 DNA/RNA 以及 DNA/DNA 探针/靶标杂交化学中都有发现。使用 T7-引物扩增样品 RNA 与 G 偏倚的强烈正幅度相关,而使用随机引物的替代扩增方案则产生小得多且部分甚至为负幅度。我们应用位置依赖的敏感性模型来分析在沿 25 个核苷酸探针序列的一个到四个相邻核苷酸的所有可能短序列基序的上下文中探针强度的具体情况。大多数较长的基序可以使用最近邻 (NN) 模型进行充分描述。相比之下,简并鸟嘌呤的连续序列需要明确考虑下一个最近邻((GGG)项)。预处理方法,如 vsn、RMA、dChip、MAS5 和 gcRMA,只能从数据中去除不充分的 G 偏倚。
位置和基序依赖的敏感性模型解释了寡核苷酸探针强度的序列效应。我们提出了一种位置依赖的 NN+GGG 混合模型来校正与含有聚 G 基序的探针相关的强度偏差。它被实现为一个基于单个芯片的基因芯片校准算法,可以在标准预处理之前的预校正步骤中应用。