Interdisciplinary Centre for Bioinformatics, Universität Leipzig, Leipzig, Germany.
PLoS One. 2009 Nov 17;4(11):e7862. doi: 10.1371/journal.pone.0007862.
Single nucleotide polymorphism (SNP) arrays are important tools widely used for genotyping and copy number estimation. This technology utilizes the specific affinity of fragmented DNA for binding to surface-attached oligonucleotide DNA probes. We analyze the variability of the probe signals of Affymetrix GeneChip SNP arrays as a function of the probe sequence to identify relevant sequence motifs which potentially cause systematic biases of genotyping and copy number estimates.
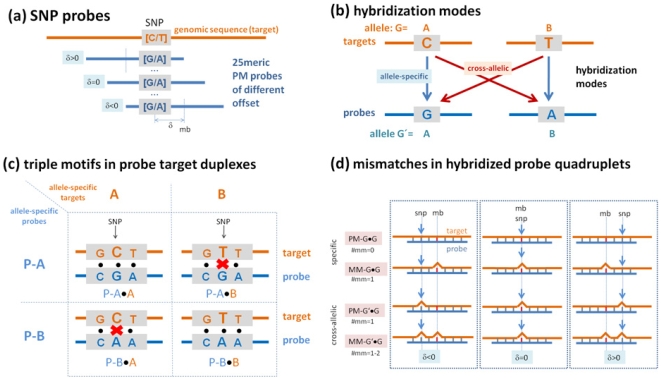
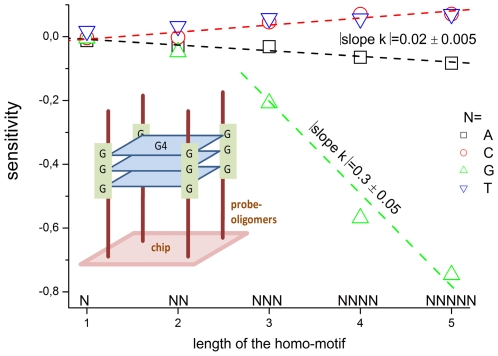
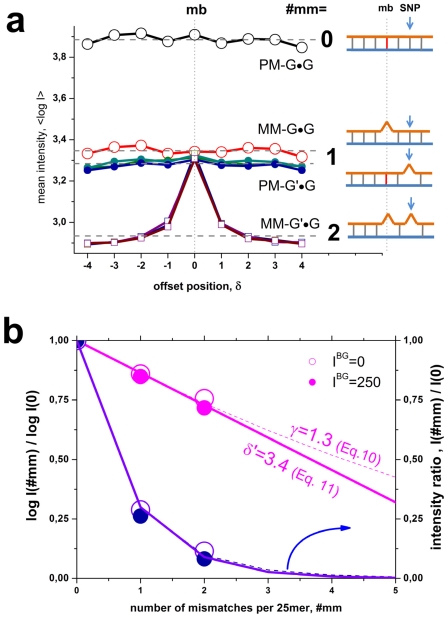
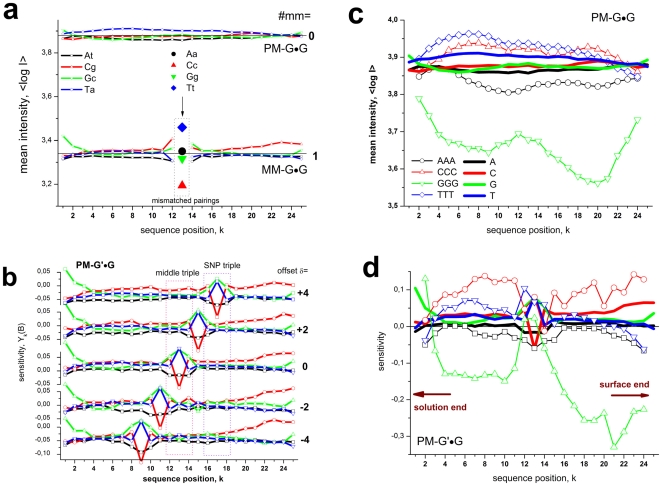
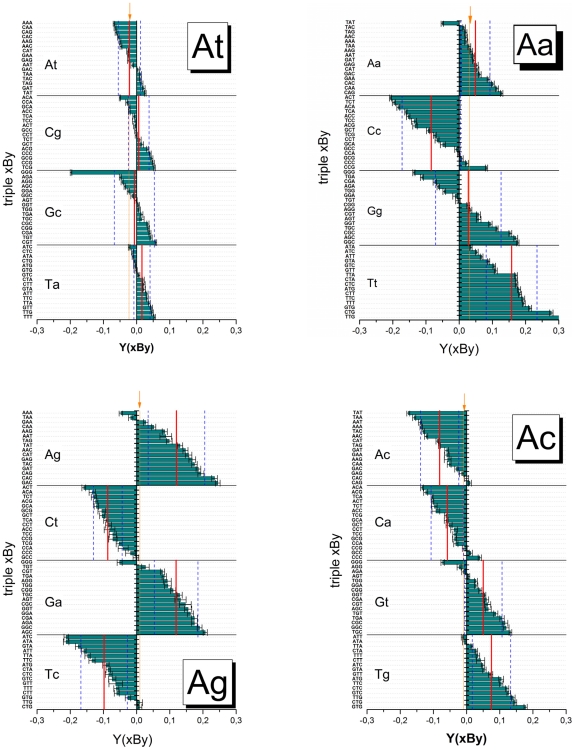
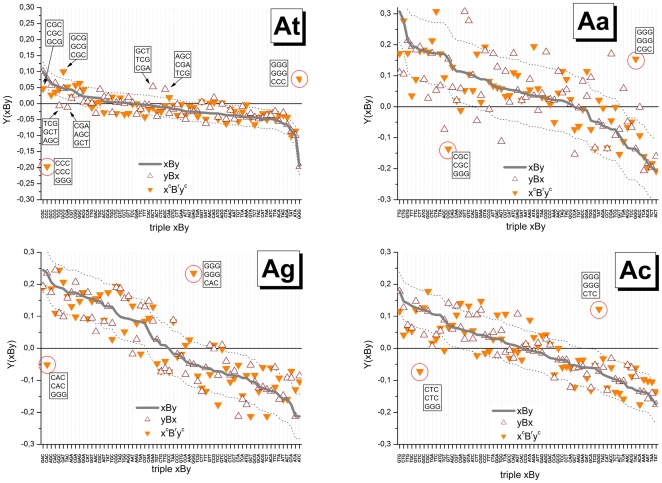
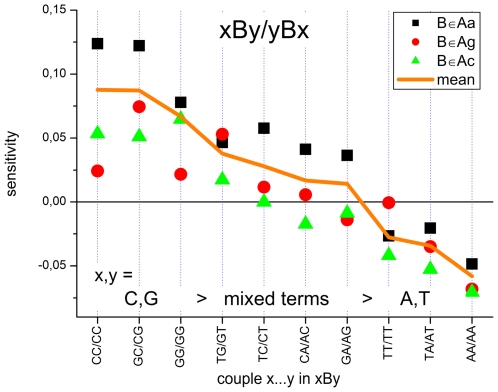
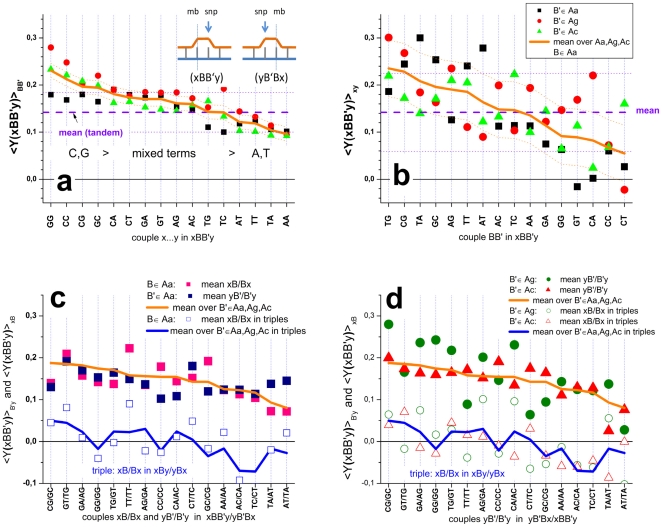
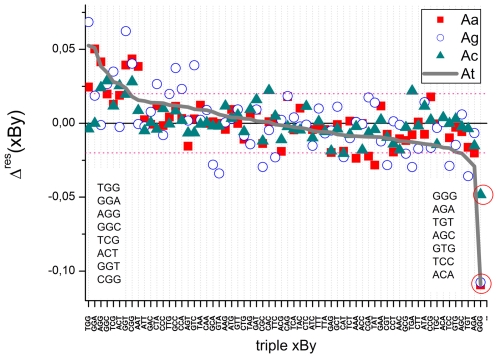
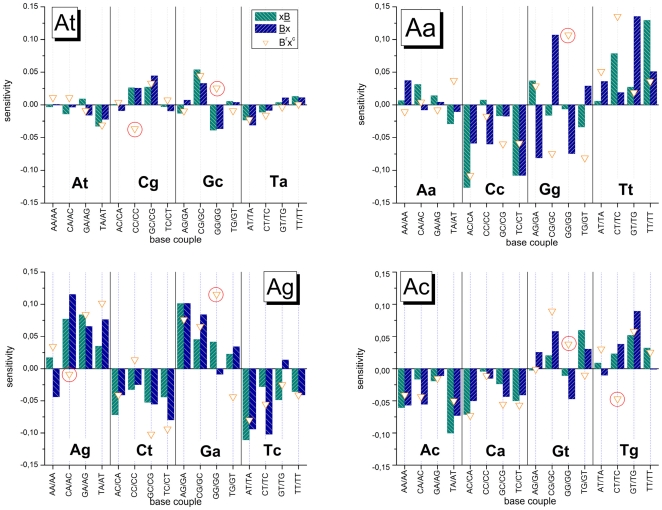
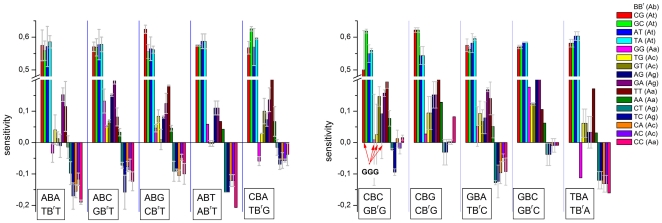
METHODOLOGY/PRINCIPAL FINDINGS: The probe design of GeneChip SNP arrays enables us to disentangle different sources of intensity modulations such as the number of mismatches per duplex, matched and mismatched base pairings including nearest and next-nearest neighbors and their position along the probe sequence. The effect of probe sequence was estimated in terms of triple-motifs with central matches and mismatches which include all 256 combinations of possible base pairings. The probe/target interactions on the chip can be decomposed into nearest neighbor contributions which correlate well with free energy terms of DNA/DNA-interactions in solution. The effect of mismatches is about twice as large as that of canonical pairings. Runs of guanines (G) and the particular type of mismatched pairings formed in cross-allelic probe/target duplexes constitute sources of systematic biases of the probe signals with consequences for genotyping and copy number estimates. The poly-G effect seems to be related to the crowded arrangement of probes which facilitates complex formation of neighboring probes with at minimum three adjacent G's in their sequence.
The applied method of "triple-averaging" represents a model-free approach to estimate the mean intensity contributions of different sequence motifs which can be applied in calibration algorithms to correct signal values for sequence effects. Rules for appropriate sequence corrections are suggested.
单核苷酸多态性(SNP)芯片是一种广泛用于基因分型和拷贝数估计的重要工具。该技术利用碎片化 DNA 与表面附着的寡核苷酸 DNA 探针结合的特定亲和力。我们分析 Affymetrix GeneChip SNP 芯片探针信号的可变性作为探针序列的函数,以确定潜在导致基因分型和拷贝数估计系统偏差的相关序列基序。
方法/主要发现:GeneChip SNP 芯片的探针设计使我们能够区分不同的强度调制源,例如每个双链体的错配数量、匹配和错配碱基对,包括最近和次近邻及其在探针序列中的位置。探针序列的影响是根据中央匹配和错配的三碱基基序来估计的,其中包括所有 256 种可能碱基配对的组合。芯片上的探针/靶标相互作用可以分解为与溶液中 DNA/DNA 相互作用的自由能项密切相关的最近邻贡献。错配的影响大约是规范配对的两倍。鸟嘌呤(G)的连续和在等位探针/靶标双链体中形成的特殊类型的错配对构成了探针信号系统偏差的来源,对基因分型和拷贝数估计有影响。聚 G 效应似乎与探针的拥挤排列有关,这种排列有利于相邻探针之间形成复杂的相互作用,在其序列中至少有三个相邻的 G。
应用的“三重平均”方法代表了一种无模型方法来估计不同序列基序的平均强度贡献,该方法可应用于校准算法中,以校正序列效应的信号值。建议了适当的序列校正规则。