Department of Pharmacology, Toxicology and Therapeutics, University of Kansas Medical Center, Kansas City, KS 66160, USA.
Toxicol Appl Pharmacol. 2010 Jul;246(1-2):8-17. doi: 10.1016/j.taap.2010.04.015. Epub 2010 Apr 25.
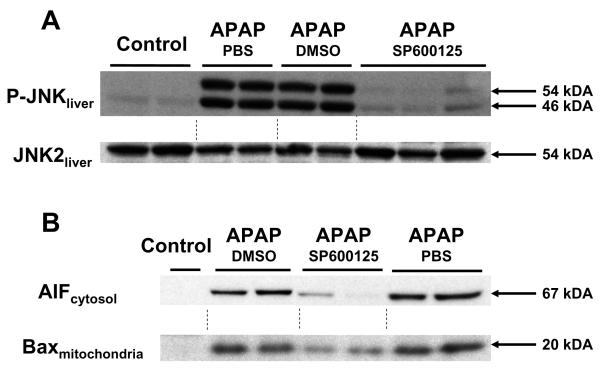
Acetaminophen (APAP) overdose, which causes liver injury in animals and humans, activates c-jun N-terminal kinase (JNK). Although it was shown that the JNK inhibitor SP600125 effectively reduced APAP hepatotoxicity, the mechanisms of protection remain unclear. C57Bl/6 mice were treated with 10mg/kg SP600125 or vehicle (8% dimethylsulfoxide) 1h before 600mg/kg APAP administration. APAP time-dependently induced JNK activation (detected by JNK phosphorylation). SP600125, but not the vehicle, reduced JNK activation, attenuated mitochondrial Bax translocation and prevented the mitochondrial release of apoptosis-inducing factor at 4-12h. Nuclear DNA fragmentation, nitrotyrosine staining, tissue GSSG levels and liver injury (plasma ALT release and necrosis) were partially attenuated by the vehicle (-65%) and completely eliminated by SP600125 (-98%) at 6 and 12h. Furthermore, SP600125 attenuated the increase of inducible nitric oxide synthase (iNOS) mRNA and protein. However, APAP did not enhance plasma nitrite+nitrate levels (NO formation); SP600125 had no effect on this parameter. The iNOS inhibitor L-NIL did not reduce NO formation or injury after APAP but prevented NO formation caused by endotoxin. Since SP600125 completely eliminated the increase in hepatic GSSG levels, an indicator of mitochondrial oxidant stress, it is concluded that the inhibition of peroxynitrite was mainly caused by reduced superoxide formation. Our data suggest that the JNK inhibitor SP600125 protects against APAP-induced liver injury in part by attenuation of mitochondrial Bax translocation but mainly by preventing mitochondrial oxidant stress and peroxynitrite formation and thereby preventing the mitochondrial permeability transition pore opening, a key event in APAP-induced cell necrosis.
对乙酰氨基酚(APAP)过量会导致动物和人类肝损伤,激活 c-jun N 端激酶(JNK)。尽管已经表明 JNK 抑制剂 SP600125 可以有效降低 APAP 肝毒性,但保护机制仍不清楚。C57Bl/6 小鼠在给予 600mg/kg APAP 前 1 小时用 10mg/kg SP600125 或载体(8%二甲基亚砜)处理。APAP 时间依赖性地诱导 JNK 激活(通过 JNK 磷酸化检测)。SP600125,但不是载体,减少 JNK 激活,减弱线粒体 Bax 易位,并防止线粒体在 4-12 小时释放凋亡诱导因子。核 DNA 片段化、硝基酪氨酸染色、组织 GSSG 水平和肝损伤(血浆 ALT 释放和坏死)在 6 和 12 小时时被载体部分减弱(-65%),并被 SP600125 完全消除(-98%)。此外,SP600125 减弱了诱导型一氧化氮合酶(iNOS)mRNA 和蛋白的增加。然而,APAP 并未增加血浆亚硝酸盐+硝酸盐水平(NO 形成);SP600125 对该参数没有影响。iNOS 抑制剂 L-NIL 不会减少 APAP 后 NO 的形成或损伤,但可防止内毒素引起的 NO 形成。由于 SP600125 完全消除了肝 GSSG 水平的增加,这是线粒体氧化剂应激的一个指标,因此可以得出结论,过氧亚硝酸盐的抑制主要是由于超氧化物形成减少所致。我们的数据表明,JNK 抑制剂 SP600125 通过减弱线粒体 Bax 易位部分保护 APAP 诱导的肝损伤,但主要通过防止线粒体氧化剂应激和过氧亚硝酸盐形成,从而防止线粒体通透性转换孔开放,这是 APAP 诱导的细胞坏死的关键事件。