Department of Pediatrics, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands.
J Inherit Metab Dis. 2010 Oct;33(5):507-11. doi: 10.1007/s10545-010-9080-z. Epub 2010 Apr 29.
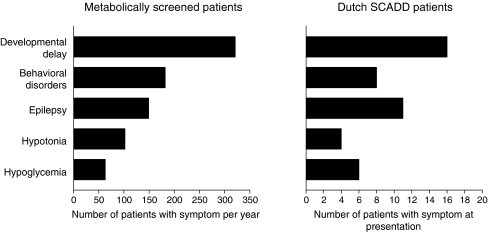
Short-chain acyl-CoA dehydrogenase deficiency (SCADD) is an autosomal recessive inborn error of mitochondrial fatty acid oxidation. SCADD is biochemically characterized by increased C4-carnitine in plasma and ethylmalonic acid in urine. The diagnosis of SCADD is confirmed by DNA analysis showing SCAD gene mutations and/or variants. SCAD gene variants are present in homozygous form in approximately 6% of the general population and considered to confer susceptibility to development of clinical disease. Clinically, SCADD generally appears to present early in life and to be most frequently associated with developmental delay, hypotonia, epilepsy, behavioral disorders, and hypoglycemia. However, these symptoms often ameliorate and even disappear spontaneously during follow-up and were found to be unrelated to the SCAD genotype. In addition, in some cases, symptoms initially attributed to SCADD could later be explained by other causes. Finally, SCADD relatives of SCADD patients as well as almost all SCADD individuals diagnosed by neonatal screening remained asymptomatic during follow-up. This potential lack of clinical consequences of SCADD has several implications. First, the diagnosis of SCADD should never preclude extension of the diagnostic workup for other potential causes of the observed symptoms. Second, patients and parents should be clearly informed about the potential lack of relevance of the disorder to avoid unfounded anxiety. Furthermore, to date, SCADD is not an optimal candidate for inclusion in newborn screening programs. More studies are needed to fully establish the relevance of SCADD and solve the question as to whether SCADD is involved in a multifactorial disease or represents a nondisease.
短链酰基辅酶 A 脱氢酶缺乏症 (SCADD) 是一种常染色体隐性遗传的线粒体脂肪酸氧化缺陷。SCADD 的生化特征是血浆中 C4-肉碱增加和尿液中乙基丙二酸增加。SCADD 的诊断通过 DNA 分析证实,显示 SCAD 基因突变和/或变体。SCAD 基因变体以纯合形式存在于约 6%的普通人群中,被认为易患临床疾病。临床上,SCADD 通常在生命早期出现,最常与发育迟缓、低张力、癫痫、行为障碍和低血糖有关。然而,这些症状通常在随访过程中会缓解甚至自行消失,并且与 SCAD 基因型无关。此外,在某些情况下,最初归因于 SCADD 的症状后来可能由其他原因解释。最后,SCADD 患者的亲属以及通过新生儿筛查诊断为 SCADD 的几乎所有个体在随访期间均无症状。SCADD 缺乏临床后果有几个含义。首先,SCADD 的诊断绝不应该排除对观察到的症状的其他潜在原因进行诊断性检查。其次,应明确告知患者和家长该疾病与症状可能缺乏相关性,以避免不必要的焦虑。此外,迄今为止,SCADD 不是新生儿筛查计划纳入的理想候选者。需要更多的研究来充分确定 SCADD 的相关性,并解决 SCADD 是否涉及多因素疾病或是否代表非疾病的问题。