School of Biology, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Bioinformatics. 2010 Aug 1;26(15):1819-26. doi: 10.1093/bioinformatics/btq284. Epub 2010 Jun 2.
New sequencing technologies have accelerated research on prokaryotic genomes and have made genome sequencing operations outside major genome sequencing centers routine. However, no off-the-shelf solution exists for the combined assembly, gene prediction, genome annotation and data presentation necessary to interpret sequencing data. The resulting requirement to invest significant resources into custom informatics support for genome sequencing projects remains a major impediment to the accessibility of high-throughput sequence data.
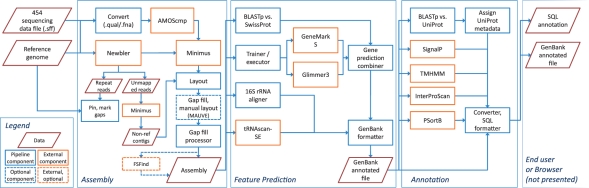
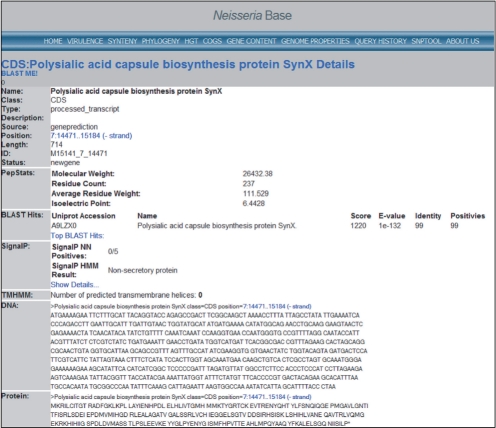
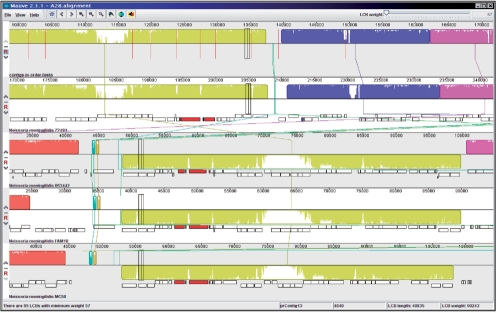
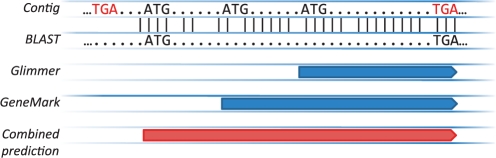
We present a self-contained, automated high-throughput open source genome sequencing and computational genomics pipeline suitable for prokaryotic sequencing projects. The pipeline has been used at the Georgia Institute of Technology and the Centers for Disease Control and Prevention for the analysis of Neisseria meningitidis and Bordetella bronchiseptica genomes. The pipeline is capable of enhanced or manually assisted reference-based assembly using multiple assemblers and modes; gene predictor combining; and functional annotation of genes and gene products. Because every component of the pipeline is executed on a local machine with no need to access resources over the Internet, the pipeline is suitable for projects of a sensitive nature. Annotation of virulence-related features makes the pipeline particularly useful for projects working with pathogenic prokaryotes.
The pipeline is licensed under the open-source GNU General Public License and available at the Georgia Tech Neisseria Base (http://nbase.biology.gatech.edu/). The pipeline is implemented with a combination of Perl, Bourne Shell and MySQL and is compatible with Linux and other Unix systems.
新的测序技术加速了对原核基因组的研究,并使主要基因组测序中心之外的基因组测序操作成为常规操作。然而,对于解释测序数据所需的组合组装、基因预测、基因组注释和数据表示,还没有现成的解决方案。因此,需要投入大量资源来为基因组测序项目提供定制的信息学支持,这仍然是高通量序列数据可及性的主要障碍。
我们提出了一个自包含的、自动化的、适用于原核测序项目的高通量开源基因组测序和计算基因组学管道。该管道已在佐治亚理工学院和疾病控制与预防中心用于分析脑膜炎奈瑟菌和支气管败血波氏杆菌基因组。该管道能够使用多种组装器和模式进行增强或手动辅助基于参考的组装;基因预测器组合;以及基因和基因产物的功能注释。由于管道的每个组件都在本地机器上执行,无需通过互联网访问资源,因此该管道适用于性质敏感的项目。与毒力相关特征的注释使该管道特别适用于与致病性原核生物合作的项目。
该管道根据开源的 GNU 通用公共许可证获得许可,并可在佐治亚理工学院的脑膜炎基地(http://nbase.biology.gatech.edu/)获得。该管道使用 Perl、Bourne Shell 和 MySQL 组合实现,与 Linux 和其他 Unix 系统兼容。