Division of Endocrinology & Metabolism, Center for Osteoporosis and Metabolic Bone Diseases, Central Arkansas Veterans Healthcare System, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
J Bone Miner Res. 2010 Nov;25(11):2427-37. doi: 10.1002/jbmr.145.
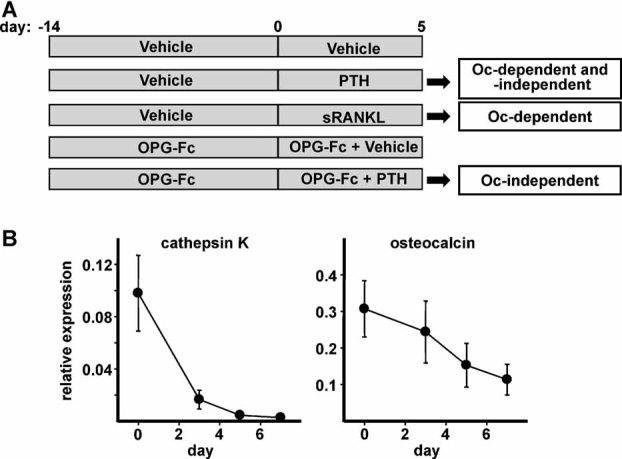
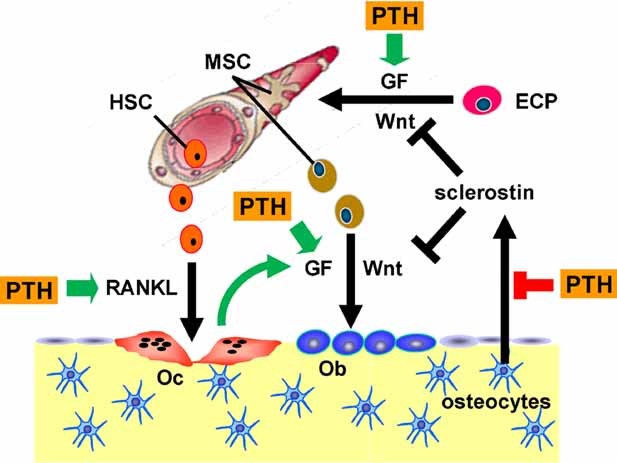
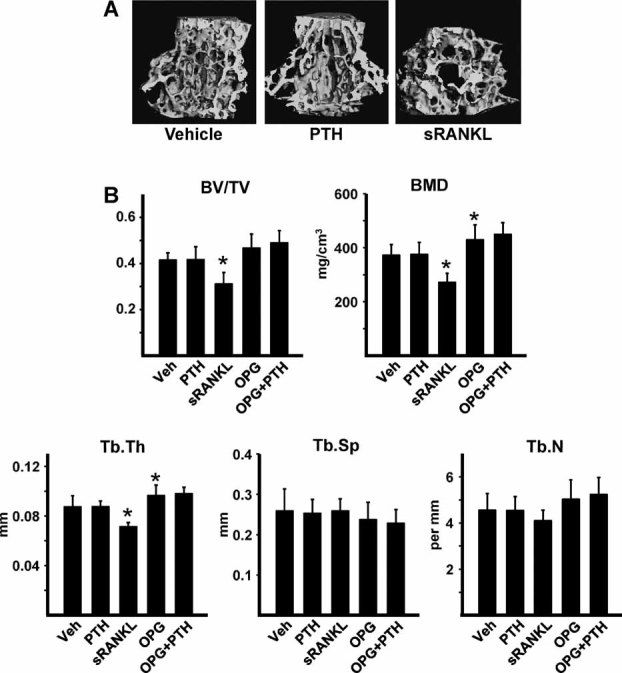
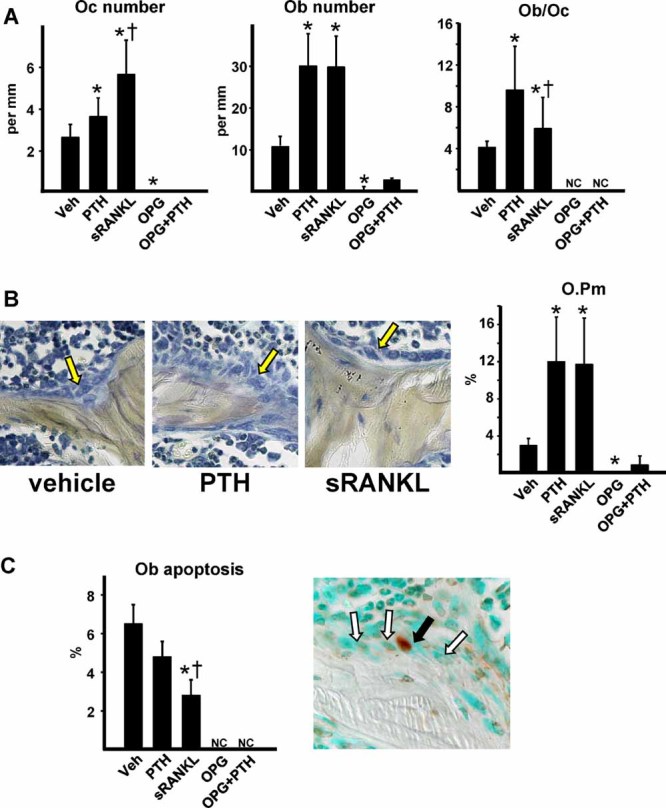
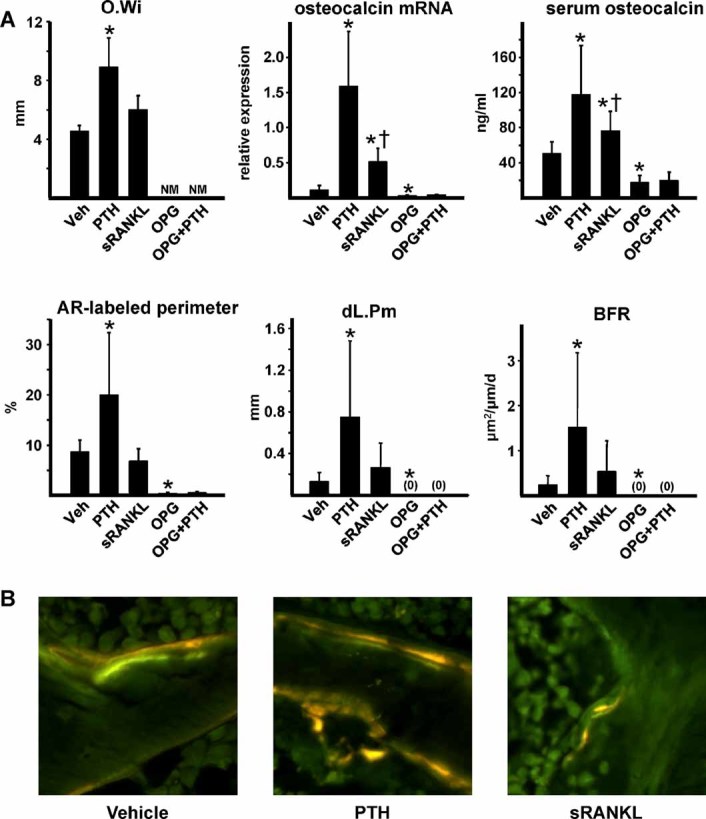
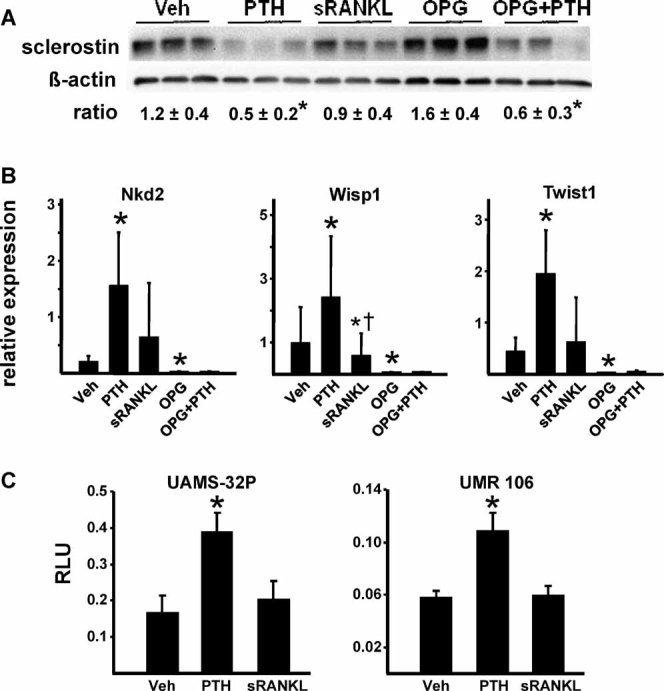
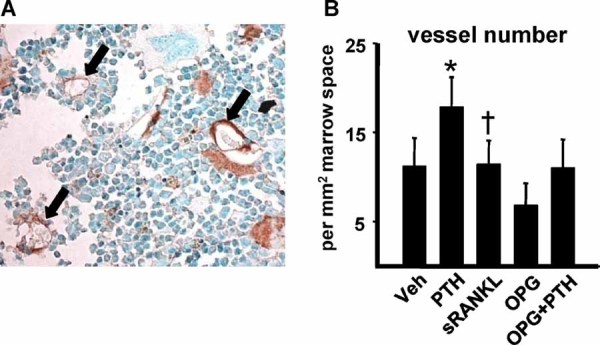
Sustained parathyroid hormone (PTH) elevation stimulates bone remodeling (ie, both resorption and formation). The former results from increased RANKL synthesis, but the cause of the latter has not been established. Current hypotheses include release of osteoblastogenic factors from osteoclasts or from the bone matrix during resorption, modulation of the production and activity of osteoblastogenic factors from cells of the osteoblast lineage, and increased angiogenesis. To dissect the contribution of these mechanisms, 6-month-old Swiss-Webster mice were infused for 5 days with 470 ng/h PTH(1-84) or 525 ng/h soluble RANKL (sRANKL). Both agents increased osteoclasts and osteoblasts in vertebral cancellous bone, but the ratio of osteoblasts to osteoclasts and the increase in bone formation was greater in PTH-treated mice. Cancellous bone mass was maintained in mice receiving PTH but lost in mice receiving sRANKL, indicating that maintenance of balanced remodeling requires osteoblastogenic effects beyond those mediated by osteoclasts. Consistent with this contention, PTH, but not sRANKL, decreased the level of the Wnt antagonist sclerostin and increased the expression of the Wnt target genes Nkd2, Wisp1, and Twist1. Furthermore, PTH, but not sRANKL, increased the number of blood vessels in the bone marrow. Weekly injections of the RANKL antagonist osteoprotegerin at 10 µg/g for 2 weeks prior to PTH infusion eliminated osteoclasts and osteoblasts and prevented the PTH-induced increase in osteoclasts, osteoblasts, and blood vessels. These results indicate that PTH stimulates osteoclast-dependent as well as osteoclast-independent (Wnt signaling) pro-osteoblastogenic pathways, both of which are required for balanced focal bone remodeling in cancellous bone.
甲状旁腺激素 (PTH) 的持续升高会刺激骨重塑(即吸收和形成)。前者是由于 RANKL 合成增加所致,但后者的原因尚未确定。目前的假设包括破骨细胞或在吸收过程中从骨基质中释放成骨细胞因子,调节成骨细胞谱系细胞中成骨细胞因子的产生和活性,以及增加血管生成。为了剖析这些机制的贡献,将 6 个月大的瑞士-韦伯斯特小鼠用 470ng/h PTH(1-84) 或 525ng/h 可溶性 RANKL(sRANKL) 输注 5 天。这两种药物都增加了椎骨松质骨中的破骨细胞和成骨细胞,但在接受 PTH 治疗的小鼠中,成骨细胞与破骨细胞的比值和骨形成的增加更大。在接受 PTH 治疗的小鼠中,松质骨量得以维持,但在接受 sRANKL 治疗的小鼠中则丢失,表明维持平衡的重塑需要超出破骨细胞介导的成骨细胞效应。这一论点与以下观点一致,即 PTH 而不是 sRANKL 降低了 Wnt 拮抗剂 sclerostin 的水平并增加了 Wnt 靶基因 Nkd2、Wisp1 和 Twist1 的表达。此外,PTH 而不是 sRANKL 增加了骨髓中的血管数量。在 PTH 输注前 2 周每周注射 10μg/g 的 RANKL 拮抗剂 osteoprotegerin,可消除破骨细胞和成骨细胞,并防止 PTH 诱导的破骨细胞、成骨细胞和血管增加。这些结果表明,PTH 刺激破骨细胞依赖性和破骨细胞非依赖性(Wnt 信号)促成骨细胞形成途径,这两者都是松质骨中平衡局灶性骨重塑所必需的。