Department of Chemical Engineering, West Virginia University College of Engineering and Mineral Resources, West Virginia University, Morgantown, WV 26506, USA.
BMC Cancer. 2010 Jun 15;10:291. doi: 10.1186/1471-2407-10-291.
Molecularly targeted drugs inhibit aberrant signaling within oncogenic pathways. Identifying the predominant pathways at work within a tumor is a key step towards tailoring therapies to the patient. Clinical samples pose significant challenges for proteomic profiling, an attractive approach for identifying predominant pathways. The objective of this study was to determine if information obtained from a limited sample (i.e., a single gel replicate) can provide insight into the predominant pathways in two well-characterized breast cancer models.
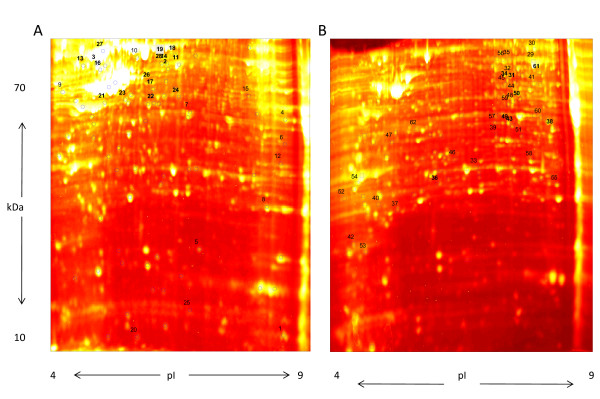
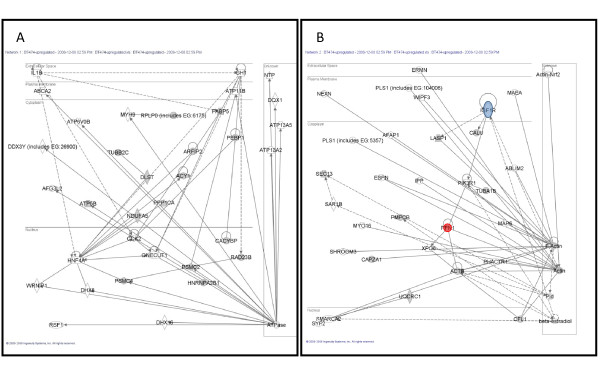
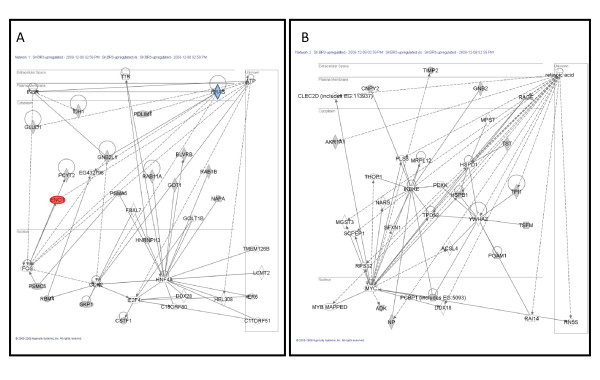
A comparative proteomic analysis of total cell lysates was obtained from two cellular models of breast cancer, BT474 (HER2+/ER+) and SKBR3 (HER2+/ER-), using two-dimensional electrophoresis and MALDI-TOF mass spectrometry. Protein interaction networks and canonical pathways were extracted from the Ingenuity Pathway Knowledgebase (IPK) based on association with the observed pattern of differentially expressed proteins.
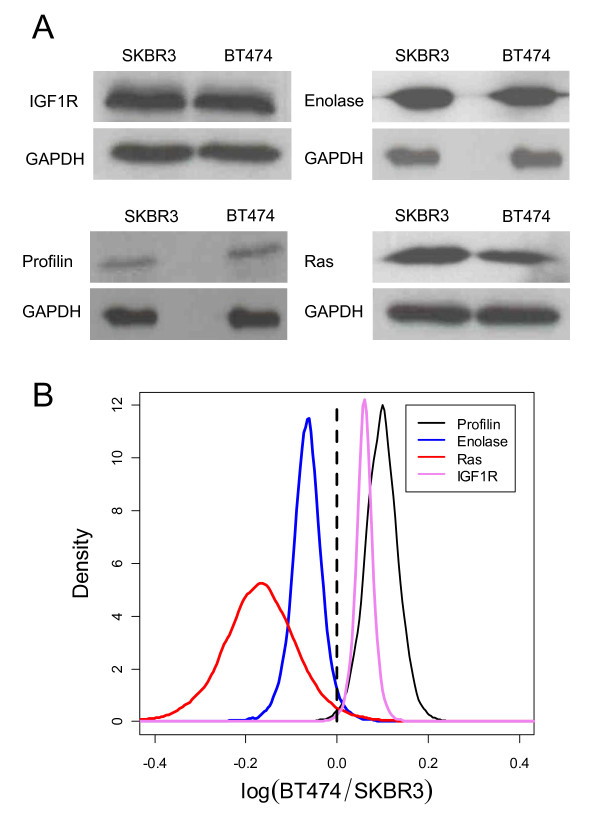
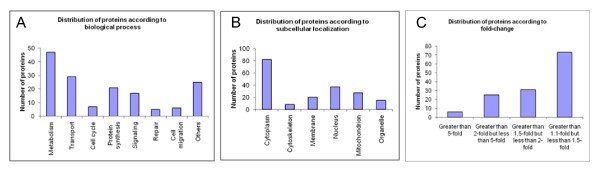
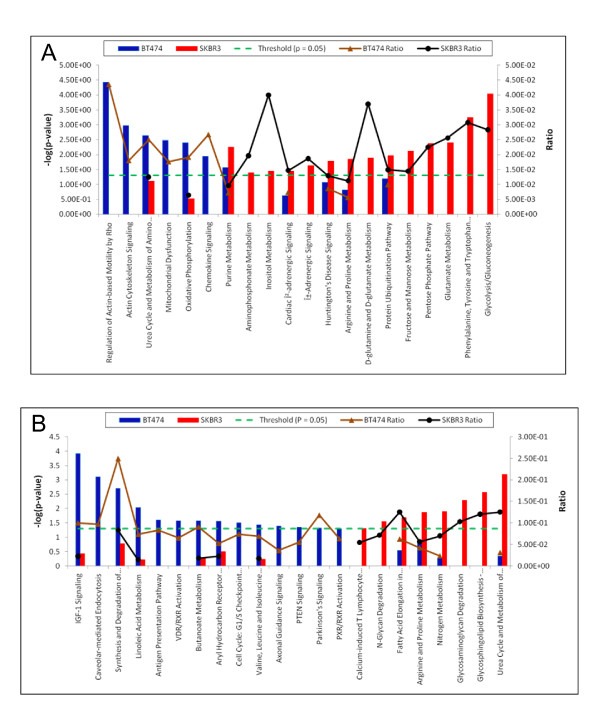
Of the 304 spots that were picked, 167 protein spots were identified. A threshold of 1.5-fold was used to select 62 proteins used in the analysis. IPK analysis suggested that metabolic pathways were highly associated with protein expression in SKBR3 cells while cell motility pathways were highly associated with BT474 cells. Inferred protein networks were confirmed by observing an up-regulation of IGF-1R and profilin in BT474 and up-regulation of Ras and enolase in SKBR3 using western blot.
When interpreted in the context of prior information, our results suggest that the overall patterns of differential protein expression obtained from limited samples can still aid in clinical decision making by providing an estimate of the predominant pathways that underpin cellular phenotype.
分子靶向药物抑制致癌途径中的异常信号。确定肿瘤中起作用的主要途径是将治疗方法针对患者的关键步骤。临床样本对蛋白质组学分析提出了重大挑战,这是识别主要途径的一种有吸引力的方法。本研究的目的是确定从有限的样本(即单个凝胶重复)中获得的信息是否可以深入了解两种经过充分表征的乳腺癌模型中的主要途径。
使用二维电泳和 MALDI-TOF 质谱法从两种乳腺癌细胞模型 BT474(HER2+/ER+)和 SKBR3(HER2+/ER-)中获得总细胞裂解物的比较蛋白质组学分析。根据与观察到的差异表达蛋白模式的关联,从 Ingenuity Pathway Knowledgebase(IPK)中提取蛋白质相互作用网络和经典途径。
在挑选的 304 个斑点中,鉴定出 167 个蛋白质斑点。使用 1.5 倍的阈值选择了用于分析的 62 种蛋白质。IPK 分析表明,代谢途径与 SKBR3 细胞中的蛋白质表达高度相关,而细胞迁移途径与 BT474 细胞高度相关。通过观察 BT474 中 IGF-1R 和原肌球蛋白的上调以及 SKBR3 中 Ras 和烯醇酶的上调,Western blot 证实了推断的蛋白质网络。
当从先前的信息背景进行解释时,我们的结果表明,从有限的样本中获得的差异蛋白表达的总体模式仍然可以通过估计支持细胞表型的主要途径来帮助临床决策。