Ho Hsiang, Milenković Tijana, Memisević Vesna, Aruri Jayavani, Przulj Natasa, Ganesan Anand K
Department of Biological Chemistry, University of California, Irvine, 92697-1700, USA.
BMC Syst Biol. 2010 Jun 15;4:84. doi: 10.1186/1752-0509-4-84.
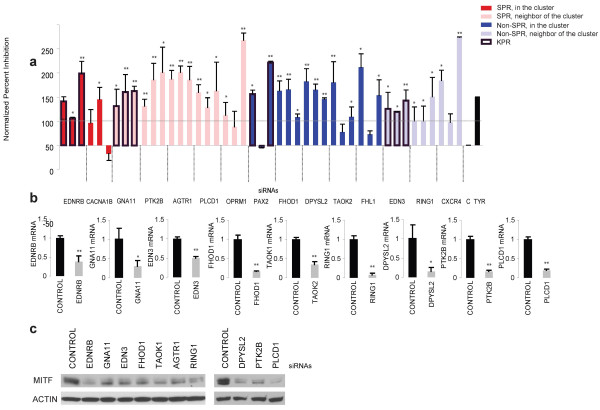
RNA-mediated interference (RNAi)-based functional genomics is a systems-level approach to identify novel genes that control biological phenotypes. Existing computational approaches can identify individual genes from RNAi datasets that regulate a given biological process. However, currently available methods cannot identify which RNAi screen "hits" are novel components of well-characterized biological pathways known to regulate the interrogated phenotype. In this study, we describe a method to identify genes from RNAi datasets that are novel components of known biological pathways. We experimentally validate our approach in the context of a recently completed RNAi screen to identify novel regulators of melanogenesis.
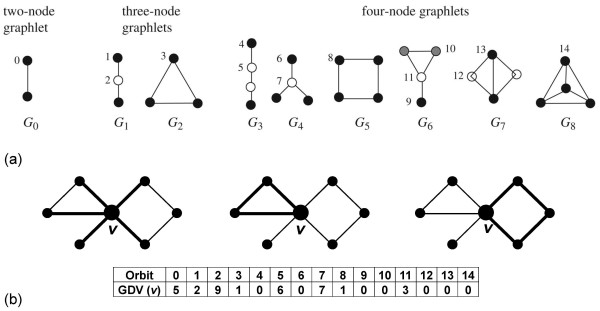
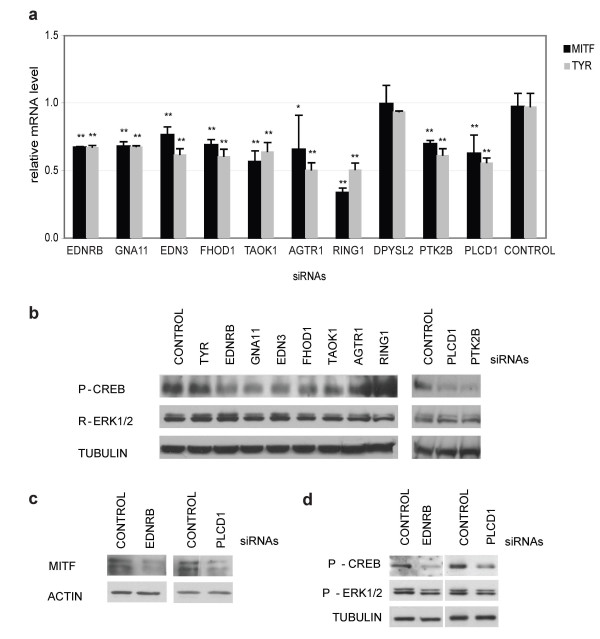
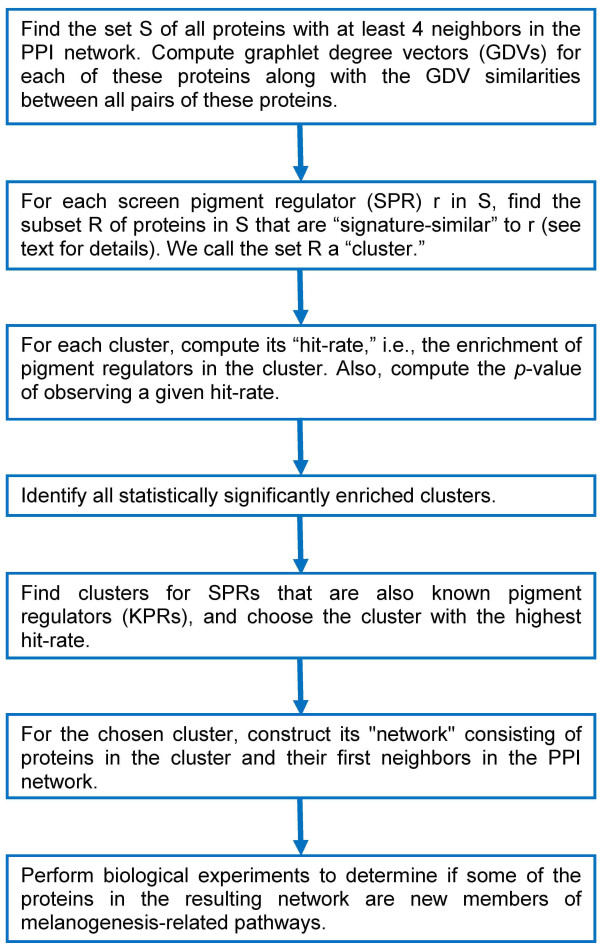
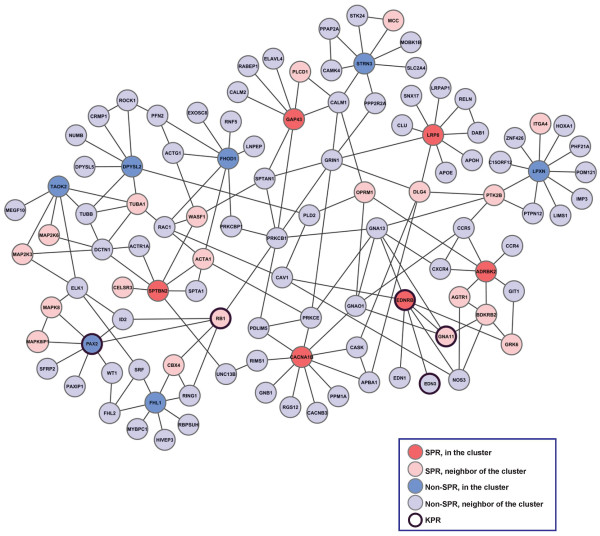
In this study, we utilize a PPI network topology-based approach to identify targets within our RNAi dataset that may be components of known melanogenesis regulatory pathways. Our computational approach identifies a set of screen targets that cluster topologically in a human PPI network with the known pigment regulator Endothelin receptor type B (EDNRB). Validation studies reveal that these genes impact pigment production and EDNRB signaling in pigmented melanoma cells (MNT-1) and normal melanocytes.
We present an approach that identifies novel components of well-characterized biological pathways from functional genomics datasets that could not have been identified by existing statistical and computational approaches.
基于RNA介导的干扰(RNAi)的功能基因组学是一种系统水平的方法,用于识别控制生物学表型的新基因。现有的计算方法可以从调节给定生物学过程的RNAi数据集中识别单个基因。然而,目前可用的方法无法确定哪些RNAi筛选“命中”是已知调节所研究表型的特征明确的生物学途径的新组成部分。在本研究中,我们描述了一种从RNAi数据集中识别作为已知生物学途径新组成部分的基因的方法。我们在最近完成的RNAi筛选的背景下通过实验验证了我们的方法,以识别黑素生成的新调节因子。
在本研究中,我们利用基于蛋白质-蛋白质相互作用(PPI)网络拓扑的方法来识别我们RNAi数据集中可能是已知黑素生成调节途径组成部分的靶点。我们的计算方法识别出一组筛选靶点,这些靶点在人类PPI网络中与已知的色素调节因子内皮素B型受体(EDNRB)在拓扑结构上聚集在一起。验证研究表明,这些基因影响色素沉着性黑色素瘤细胞(MNT-1)和正常黑素细胞中的色素产生和EDNRB信号传导。
我们提出了一种方法,可从功能基因组学数据集中识别特征明确的生物学途径的新组成部分,而现有统计和计算方法无法识别这些组成部分。