J. Craig Venter Institute, Rockville, Maryland, United States of America.
PLoS Negl Trop Dis. 2010 Jun 15;4(6):e716. doi: 10.1371/journal.pntd.0000716.
In order to maintain genome information accurately and relevantly, original genome annotations need to be updated and evaluated regularly. Manual reannotation of genomes is important as it can significantly reduce the propagation of errors and consequently diminishes the time spent on mistaken research. For this reason, after five years from the initial submission of the Entamoeba histolytica draft genome publication, we have re-examined the original 23 Mb assembly and the annotation of the predicted genes.
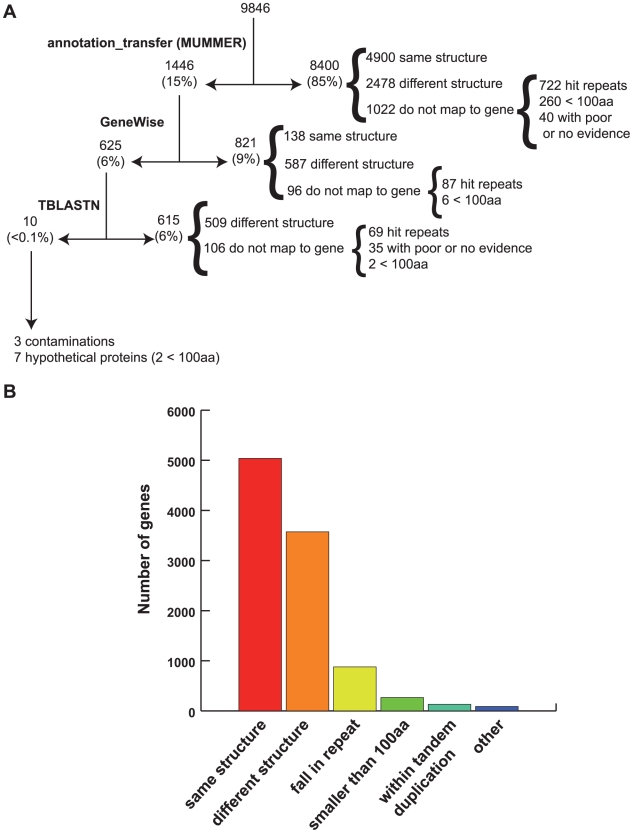
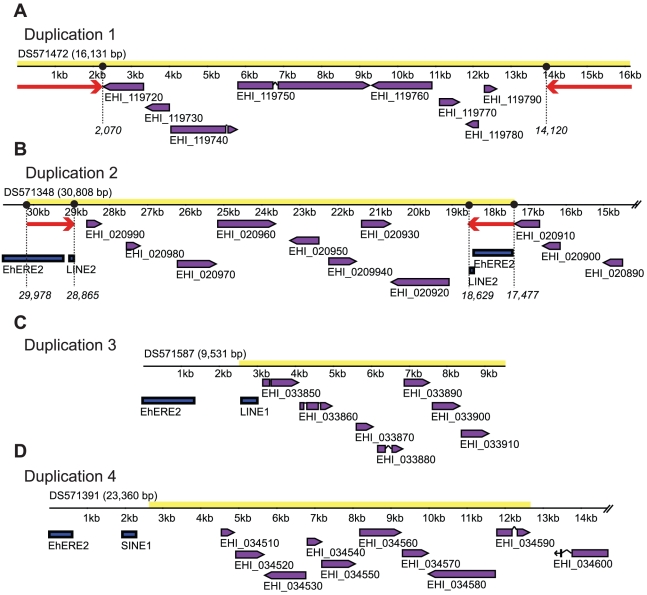
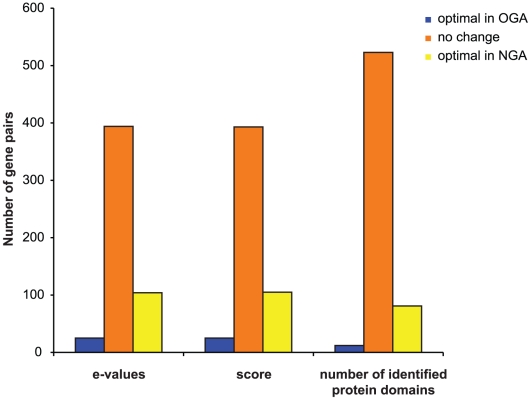
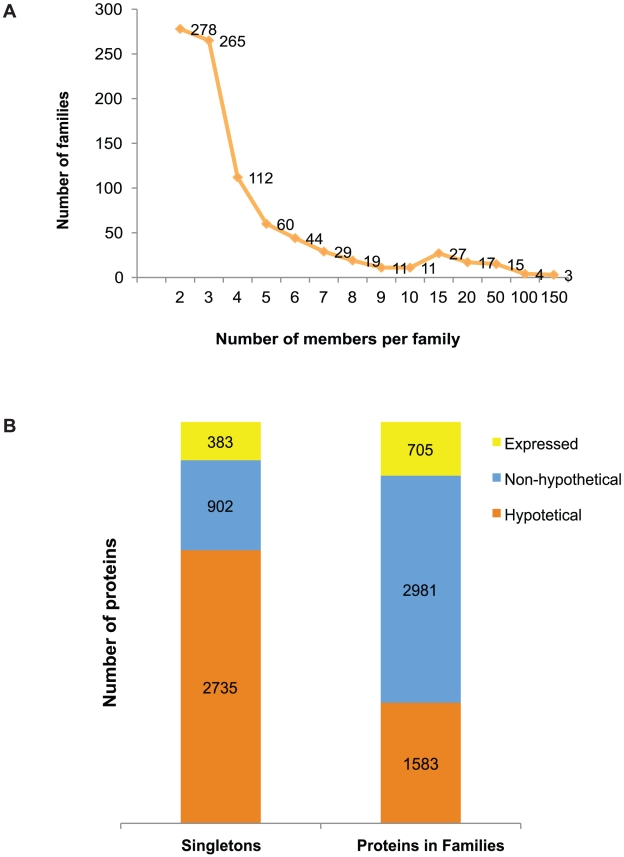
The evaluation of the genomic sequence led to the identification of more than one hundred artifactual tandem duplications that were eliminated by re-assembling the genome. The reannotation was done using a combination of manual and automated genome analysis. The new 20 Mb assembly contains 1,496 scaffolds and 8,201 predicted genes, of which 60% are identical to the initial annotation and the remaining 40% underwent structural changes. Functional classification of 60% of the genes was modified based on recent sequence comparisons and new experimental data. We have assigned putative function to 3,788 proteins (46% of the predicted proteome) based on the annotation of predicted gene families, and have identified 58 protein families of five or more members that share no homology with known proteins and thus could be entamoeba specific. Genome analysis also revealed new features such as the presence of segmental duplications of up to 16 kb flanked by inverted repeats, and the tight association of some gene families with transposable elements.
This new genome annotation and analysis represents a more refined and accurate blueprint of the pathogen genome, and provides an upgraded tool as reference for the study of many important aspects of E. histolytica biology, such as genome evolution and pathogenesis.
为了准确和相关地维护基因组信息,需要定期更新和评估原始基因组注释。对基因组进行手动重新注释很重要,因为它可以显著减少错误的传播,从而减少在错误研究上花费的时间。出于这个原因,在初始提交溶组织内阿米巴虫草案基因组出版物五年后,我们重新检查了原始的 23Mb 组装和预测基因的注释。
对基因组序列的评估导致鉴定了一百多个人为的串联重复,这些重复通过重新组装基因组而被消除。重新注释是使用手动和自动化基因组分析相结合的方法完成的。新的 20Mb 组装包含 1496 个支架和 8201 个预测基因,其中 60%与初始注释相同,其余 40%发生了结构变化。根据最近的序列比较和新的实验数据,对 60%的基因进行了功能分类修改。我们根据预测基因家族的注释,为 3788 个蛋白质(预测蛋白质组的 46%)分配了可能的功能,并确定了 58 个具有五个或更多成员的蛋白质家族,这些家族与已知蛋白质没有同源性,因此可能是内阿米巴虫特有的。基因组分析还揭示了新的特征,例如长达 16kb 的片段重复,侧翼为反向重复,以及一些基因家族与转座元件的紧密关联。
这个新的基因组注释和分析代表了病原体基因组更精细和准确的蓝图,并提供了一个升级的工具作为参考,用于研究许多重要的溶组织内阿米巴虫生物学方面,如基因组进化和发病机制。