Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA 92093, USA.
Chem Biol Drug Des. 2010 Sep 1;76(3):201-17. doi: 10.1111/j.1747-0285.2010.01012.x. Epub 2010 Jul 5.
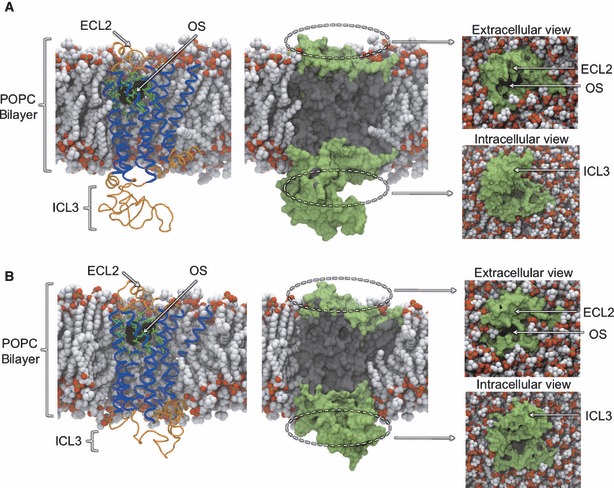
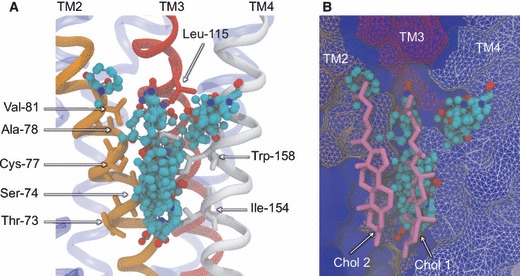
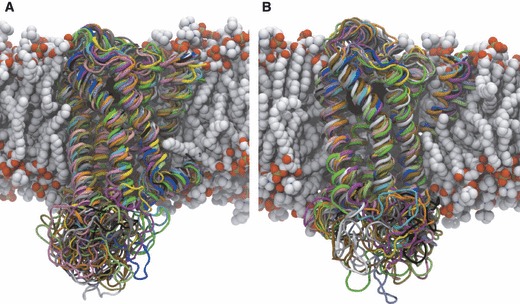
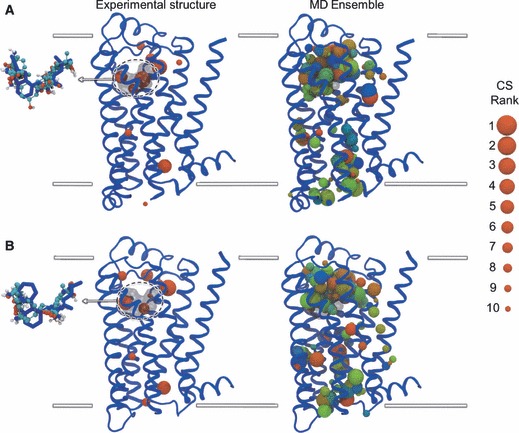
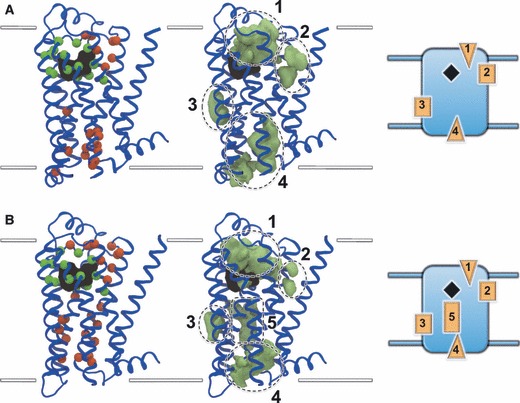
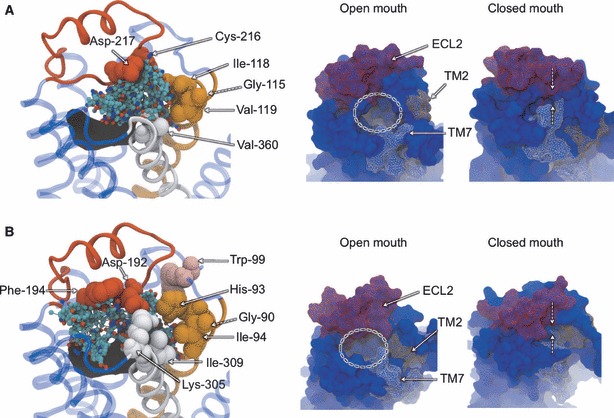
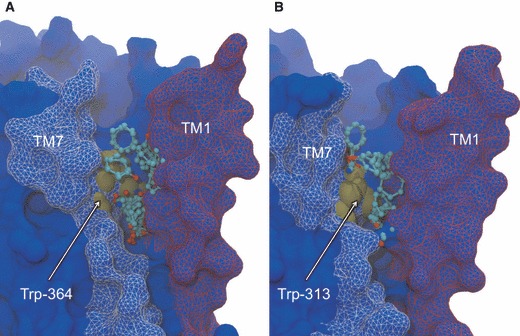
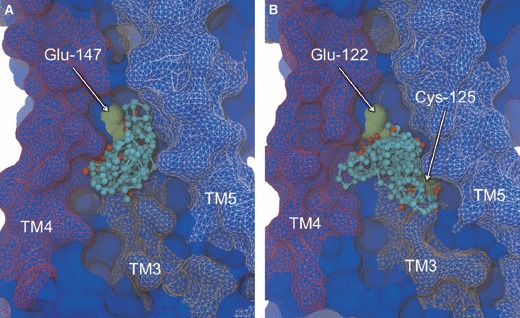
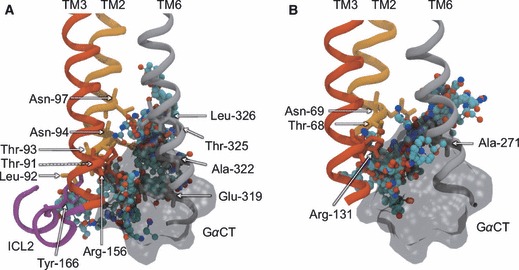
To address the problem of specificity in G-protein coupled receptor (GPCR) drug discovery, there has been tremendous recent interest in allosteric drugs that bind at sites topographically distinct from the orthosteric site. Unfortunately, structure-based drug design of allosteric GPCR ligands has been frustrated by the paucity of structural data for allosteric binding sites, making a strong case for predictive computational methods. In this work, we map the surfaces of the beta1 (beta1AR) and beta2 (beta2AR) adrenergic receptor structures to detect a series of five potentially druggable allosteric sites. We employ the FTMAP algorithm to identify 'hot spots' with affinity for a variety of organic probe molecules corresponding to drug fragments. Our work is distinguished by an ensemble-based approach, whereby we map diverse receptor conformations taken from molecular dynamics (MD) simulations totaling approximately 0.5 micros. Our results reveal distinct pockets formed at both solvent-exposed and lipid-exposed cavities, which we interpret in light of experimental data and which may constitute novel targets for GPCR drug discovery. This mapping data can now serve to drive a combination of fragment-based and virtual screening approaches for the discovery of small molecules that bind at these sites and which may offer highly selective therapies.
为了解决 G 蛋白偶联受体 (GPCR) 药物发现中特异性的问题,人们最近对结合在变构部位而非正位部位的变构药物产生了浓厚的兴趣。不幸的是,由于变构结合部位的结构数据稀缺,基于结构的变构 GPCR 配体药物设计受到了阻碍,这强烈需要预测性计算方法。在这项工作中,我们将β1(β1AR)和β2(β2AR)肾上腺素能受体结构的表面进行映射,以检测一系列五个潜在可成药的变构部位。我们采用 FTMAP 算法来识别与各种有机探针分子(对应于药物片段)具有亲和力的“热点”。我们的工作的特点是采用基于集合的方法,我们从分子动力学(MD)模拟中映射了来自不同受体构象的集合,模拟总时长约为 0.5 微秒。我们的结果揭示了在溶剂暴露和脂质暴露腔中形成的不同口袋,我们根据实验数据对其进行了解释,这些口袋可能构成 GPCR 药物发现的新靶标。现在,这些映射数据可用于驱动基于片段和虚拟筛选方法的小分子发现,这些小分子可以结合这些部位,并提供高度选择性的治疗方法。