Department of Biology of Namur University, Belgium.
BMC Bioinformatics. 2010 Jul 15;11:379. doi: 10.1186/1471-2105-11-379.
The development, in the last decade, of stochastic heuristics implemented in robust application softwares has made large phylogeny inference a key step in most comparative studies involving molecular sequences. Still, the choice of a phylogeny inference software is often dictated by a combination of parameters not related to the raw performance of the implemented algorithm(s) but rather by practical issues such as ergonomics and/or the availability of specific functionalities.
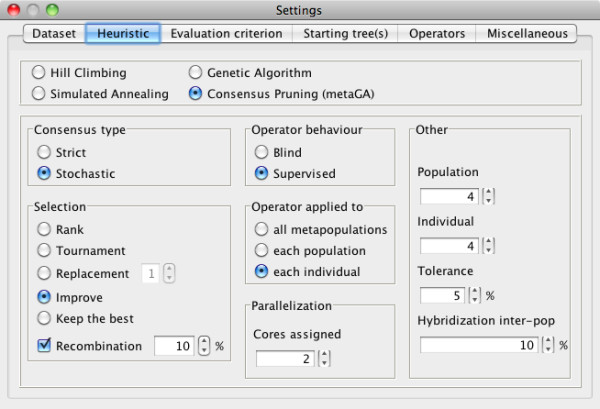
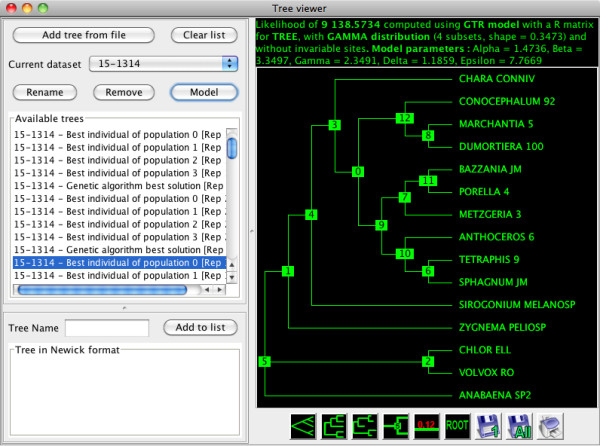
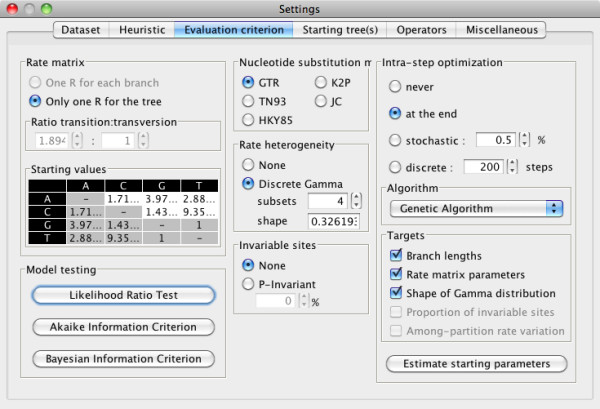
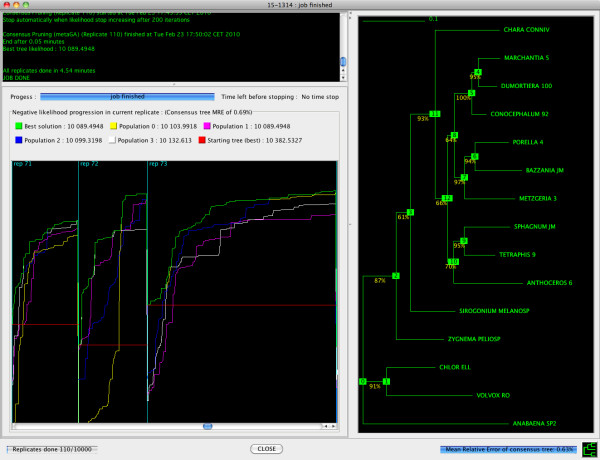
Here, we present MetaPIGA v2.0, a robust implementation of several stochastic heuristics for large phylogeny inference (under maximum likelihood), including a Simulated Annealing algorithm, a classical Genetic Algorithm, and the Metapopulation Genetic Algorithm (metaGA) together with complex substitution models, discrete Gamma rate heterogeneity, and the possibility to partition data. MetaPIGA v2.0 also implements the Likelihood Ratio Test, the Akaike Information Criterion, and the Bayesian Information Criterion for automated selection of substitution models that best fit the data. Heuristics and substitution models are highly customizable through manual batch files and command line processing. However, MetaPIGA v2.0 also offers an extensive graphical user interface for parameters setting, generating and running batch files, following run progress, and manipulating result trees. MetaPIGA v2.0 uses standard formats for data sets and trees, is platform independent, runs in 32 and 64-bits systems, and takes advantage of multiprocessor and multicore computers.
The metaGA resolves the major problem inherent to classical Genetic Algorithms by maintaining high inter-population variation even under strong intra-population selection. Implementation of the metaGA together with additional stochastic heuristics into a single software will allow rigorous optimization of each heuristic as well as a meaningful comparison of performances among these algorithms. MetaPIGA v2.0 gives access both to high customization for the phylogeneticist, as well as to an ergonomic interface and functionalities assisting the non-specialist for sound inference of large phylogenetic trees using nucleotide sequences. MetaPIGA v2.0 and its extensive user-manual are freely available to academics at http://www.metapiga.org.
在过去十年中,稳健应用软件中开发的随机启发式算法使得大规模系统发育推断成为大多数涉及分子序列的比较研究的关键步骤。然而,系统发育推断软件的选择通常取决于多种参数,这些参数与所实现算法的原始性能无关,而是与实际问题(如人体工程学和/或特定功能的可用性)有关。
这里,我们介绍了 MetaPIGA v2.0,这是一种用于大规模系统发育推断(最大似然法)的多种随机启发式算法的稳健实现,包括模拟退火算法、经典遗传算法和 Metapopulation Genetic Algorithm(metaGA),以及复杂的替代模型、离散 Gamma 率异质性,以及数据分区的可能性。MetaPIGA v2.0 还实现了似然比检验、Akaike 信息准则和贝叶斯信息准则,用于自动选择最适合数据的替代模型。启发式和替代模型可以通过手动批处理文件和命令行处理进行高度定制。然而,MetaPIGA v2.0 还为参数设置、生成和运行批处理文件、跟踪运行进度以及操作结果树提供了广泛的图形用户界面。MetaPIGA v2.0 使用标准格式的数据和树,独立于平台,可在 32 位和 64 位系统上运行,并利用多处理器和多核计算机。
metaGA 通过在强种群内选择下保持高种群间变异性,解决了经典遗传算法固有的主要问题。将 metaGA 与其他随机启发式算法集成到单个软件中,将允许对每个启发式算法进行严格优化,并对这些算法的性能进行有意义的比较。MetaPIGA v2.0 为系统发育学家提供了高度定制的功能,以及一个符合人体工程学的界面和功能,帮助非专业人士使用核苷酸序列对大型系统发育树进行合理推断。MetaPIGA v2.0 及其广泛的用户手册可在 http://www.metapiga.org 上免费提供给学术界。