TCM Group, Cavendish Laboratory, University of Cambridge, Cambridge, United Kingdom.
PLoS Comput Biol. 2010 Aug 12;6(8):e1000880. doi: 10.1371/journal.pcbi.1000880.
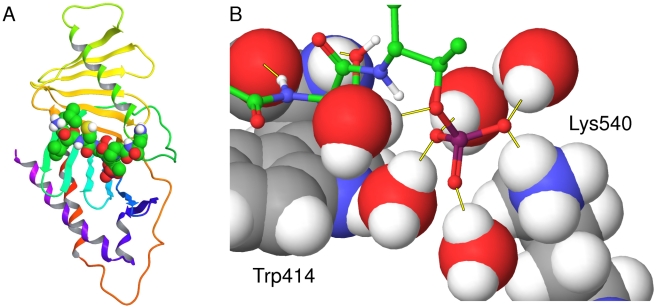
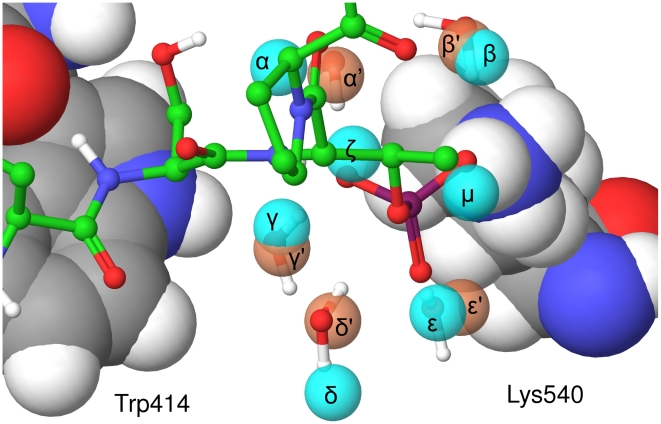
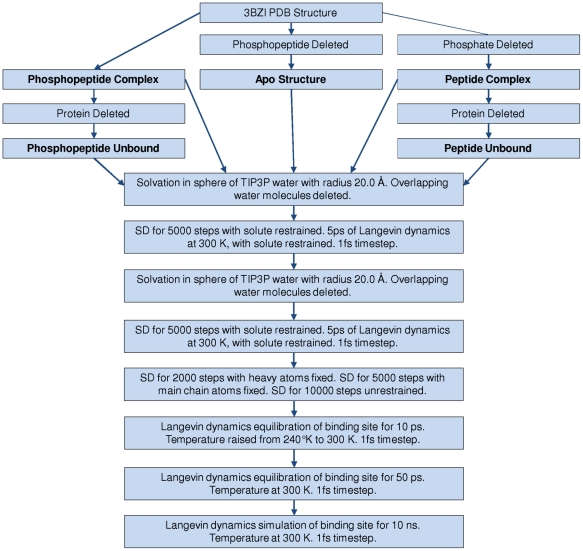
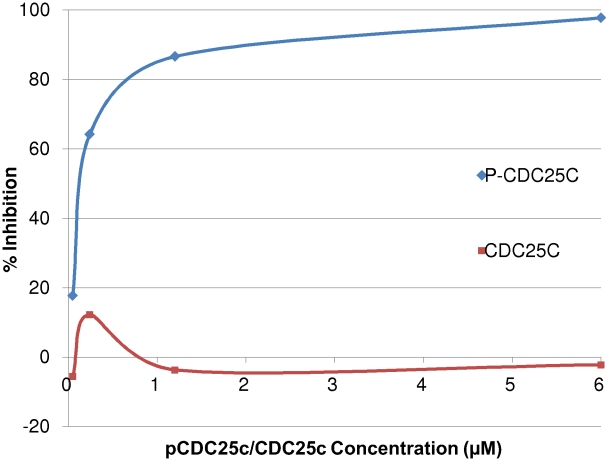
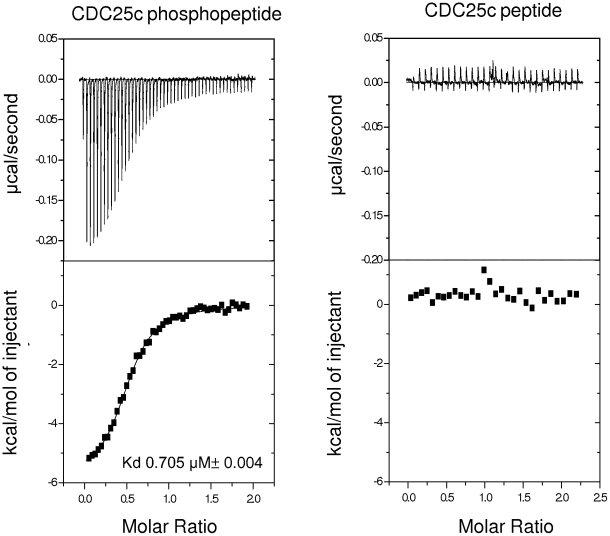
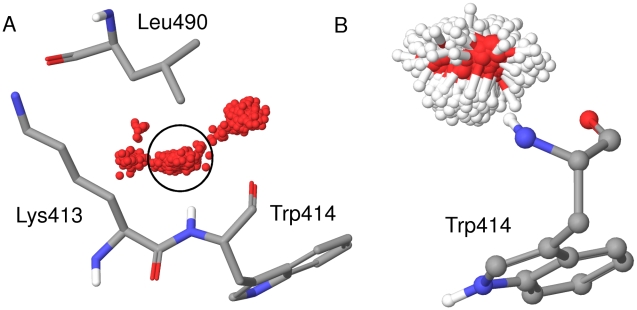
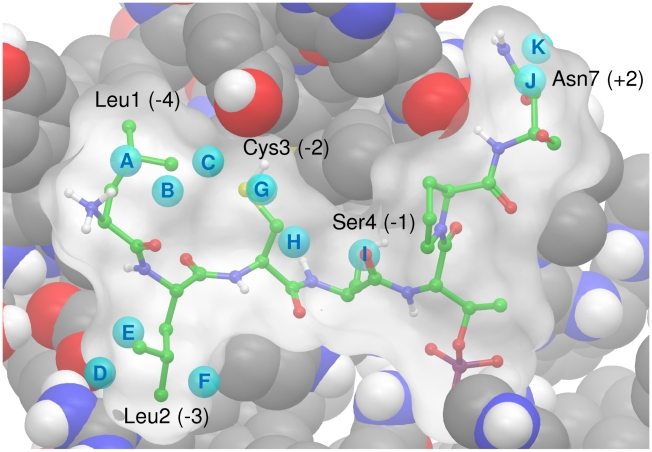
The Polo-Like Kinase 1 (PLK1) acts as a central regulator of mitosis and is over-expressed in a wide range of human tumours where high levels of expression correlate with a poor prognosis. PLK1 comprises two structural elements, a kinase domain and a polo-box domain (PBD). The PBD binds phosphorylated substrates to control substrate phosphorylation by the kinase domain. Although the PBD preferentially binds to phosphopeptides, it has a relatively broad sequence specificity in comparison with other phosphopeptide binding domains. We analysed the molecular determinants of recognition by performing molecular dynamics simulations of the PBD with one of its natural substrates, CDC25c. Predicted binding free energies were calculated using a molecular mechanics, Poisson-Boltzmann surface area approach. We calculated the per-residue contributions to the binding free energy change, showing that the phosphothreonine residue and the mainchain account for the vast majority of the interaction energy. This explains the very broad sequence specificity with respect to other sidechain residues. Finally, we considered the key role of bridging water molecules at the binding interface. We employed inhomogeneous fluid solvation theory to consider the free energy of water molecules on the protein surface with respect to bulk water molecules. Such an analysis highlights binding hotspots created by elimination of water molecules from hydrophobic surfaces. It also predicts that a number of water molecules are stabilized by the presence of the charged phosphate group, and that this will have a significant effect on the binding affinity. Our findings suggest a molecular rationale for the promiscuous binding of the PBD and highlight a role for bridging water molecules at the interface. We expect that this method of analysis will be very useful for probing other protein surfaces to identify binding hotspots for natural binding partners and small molecule inhibitors.
Polo 样激酶 1(PLK1)作为有丝分裂的中央调节因子,在广泛的人类肿瘤中过度表达,高水平的表达与预后不良相关。PLK1 由两个结构元件组成,一个激酶结构域和一个 polo 盒结构域(PBD)。PBD 结合磷酸化底物,以控制激酶结构域对底物的磷酸化。尽管 PBD 优先结合磷酸肽,但与其他磷酸肽结合结构域相比,它具有相对较宽的序列特异性。我们通过对 PBD 与其天然底物之一 CDC25c 进行分子动力学模拟来分析识别的分子决定因素。使用分子力学泊松-玻尔兹曼表面积方法计算预测的结合自由能。我们计算了结合自由能变化的每个残基贡献,结果表明磷酸苏氨酸残基和主链占相互作用能的绝大部分。这解释了相对于其他侧链残基非常广泛的序列特异性。最后,我们考虑了结合界面上桥接水分子的关键作用。我们采用非均匀流体溶剂化理论来考虑蛋白质表面上水分子相对于体相水分子的自由能。这种分析突出了通过从疏水面消除水分子而产生的结合热点。它还预测了许多水分子通过存在带电荷的磷酸基团而稳定,这将对结合亲和力产生重大影响。我们的研究结果为 PBD 的混杂结合提供了分子依据,并强调了界面上桥接水分子的作用。我们预计这种分析方法将非常有助于探测其他蛋白质表面,以识别天然结合伴侣和小分子抑制剂的结合热点。