Department of Epidemiology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA.
BMC Bioinformatics. 2010 Sep 1;11:442. doi: 10.1186/1471-2105-11-442.
Forward-time simulations have unique advantages in power and flexibility for the simulation of genetic samples of complex human diseases because they can closely mimic the evolution of human populations carrying these diseases. However, a number of methodological and computational constraints have prevented the power of this simulation method from being fully explored in existing forward-time simulation methods.
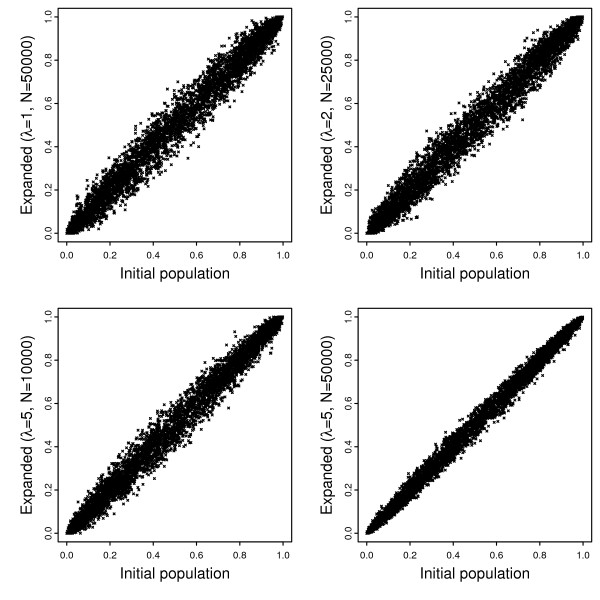
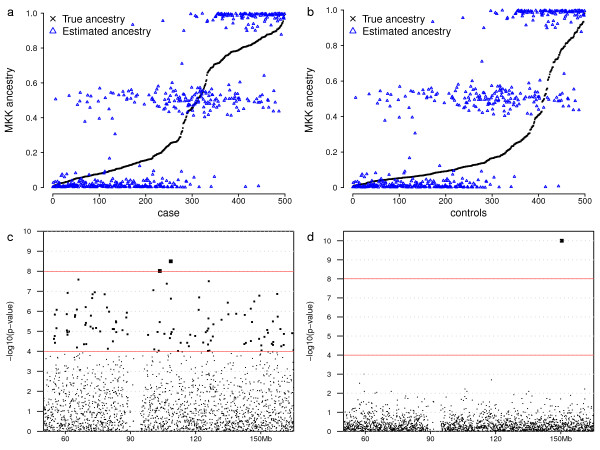
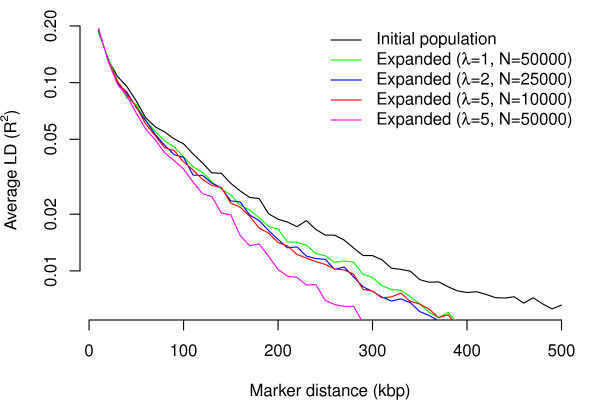
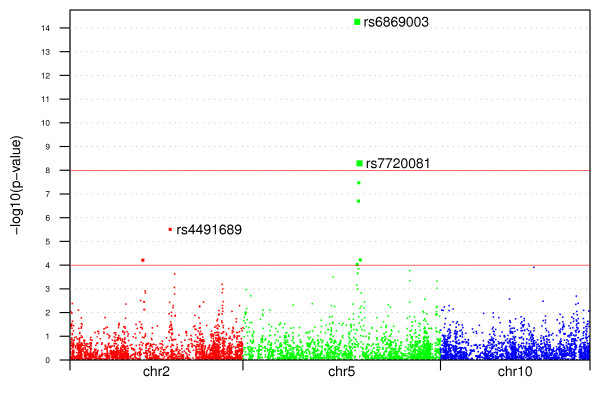
Using a general-purpose forward-time population genetics simulation environment, we developed a forward-time simulation method that can be used to simulate realistic samples for genome-wide association studies. We examined the properties of this simulation method by comparing simulated samples with real data and demonstrated its wide applicability using four examples, including a simulation of case-control samples with a disease caused by multiple interacting genetic and environmental factors, a simulation of trio families affected by a disease-predisposing allele that had been subjected to either slow or rapid selective sweep, and a simulation of a structured population resulting from recent population admixture.
Our algorithm simulates populations that closely resemble the complex structure of the human genome, while allows the introduction of signals of natural selection. Because of its flexibility to generate different types of samples with arbitrary disease or quantitative trait models, this simulation method can simulate realistic samples to evaluate the performance of a wide variety of statistical gene mapping methods for genome-wide association studies.
正向时间模拟在模拟复杂人类疾病的遗传样本方面具有独特的优势,因为它们可以紧密模拟携带这些疾病的人类群体的进化。然而,由于一些方法学和计算方面的限制,现有的正向时间模拟方法并未充分探索这种模拟方法的优势。
我们使用通用的正向时间群体遗传学模拟环境,开发了一种正向时间模拟方法,可用于模拟全基因组关联研究的真实样本。我们通过将模拟样本与真实数据进行比较来检验这种模拟方法的特性,并通过四个示例展示了其广泛的适用性,包括由多个相互作用的遗传和环境因素引起的疾病的病例对照样本的模拟、受到缓慢或快速选择压力的疾病易感等位基因的三核苷酸家庭的模拟,以及由近期群体混合引起的结构群体的模拟。
我们的算法模拟的群体与人类基因组的复杂结构非常相似,同时允许引入自然选择的信号。由于其灵活性,可以生成具有任意疾病或数量性状模型的不同类型的样本,因此这种模拟方法可以模拟真实样本,以评估全基因组关联研究中各种统计基因映射方法的性能。