Metabolic Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bldg. 10 Room 8C-101, 10 Center Dr. MSC 1752 Bethesda, MD 20892-1752, USA.
Best Pract Res Clin Endocrinol Metab. 2010 Jun;24(3):491-502. doi: 10.1016/j.beem.2010.01.003.
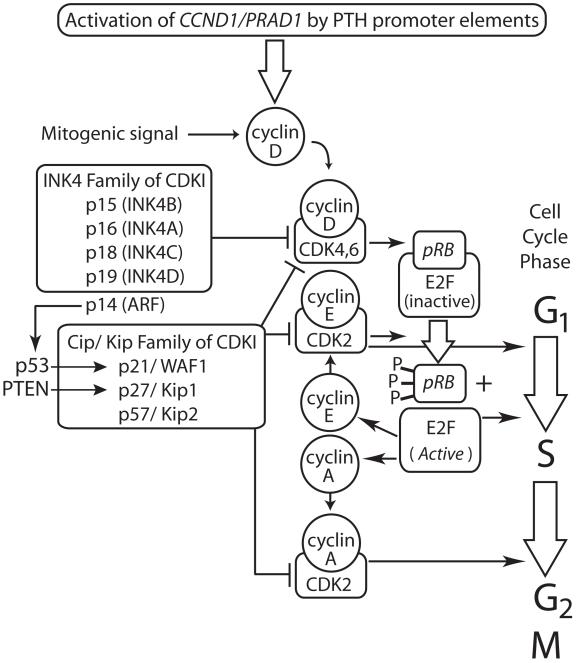
Primary hyperparathyroidism (HPT) results from the excessive secretion of parathyroid hormone from parathyroid tumours. While most HPT is sporadic, it is associated with a familial syndrome in a minority of cases. The study of these syndromes has helped define the pathophysiology of both familial and sporadic parathyroid neoplasms. Investigation of kindred with multiple endocrine neoplasia type 1 (MEN1) and the hyperparathyroidism-jaw tumour syndrome (HPT-JT) led to the discovery of the tumour suppressor genes MEN1 and HRPT2. We now recognise that somatic mutations in MEN1 and HRPT2 tumour suppressor genes are frequent events in sporadic parathyroid adenomas and carcinomas, respectively. Parathyroid tumours in the MEN2A syndrome result from mutational activation of the RET oncogene. The CCND1/PRAD1 oncogene was discovered by analysis of sporadic parathyroid tumours. Studies of familial isolated HPT and analysis of chromosomal loss and gain in parathyroid tumours suggest that other genes relevant to parathyroid neoplasia await identification.
原发性甲状旁腺功能亢进症(HPT)是由于甲状旁腺肿瘤过度分泌甲状旁腺激素引起的。虽然大多数 HPT 是散发性的,但在少数情况下与家族性综合征有关。对这些综合征的研究有助于阐明家族性和散发性甲状旁腺肿瘤的病理生理学。对多发性内分泌腺瘤 1 型(MEN1)和甲状旁腺功能亢进-颌骨肿瘤综合征(HPT-JT)家族的研究导致了肿瘤抑制基因 MEN1 和 HRPT2 的发现。我们现在认识到,MEN1 和 HRPT2 肿瘤抑制基因的体细胞突变分别是散发性甲状旁腺腺瘤和癌的常见事件。MEN2A 综合征中的甲状旁腺肿瘤是由于 RET 癌基因的突变激活引起的。CCND1/PRAD1 癌基因是通过对散发性甲状旁腺肿瘤的分析发现的。家族性孤立性 HPT 的研究以及甲状旁腺肿瘤的染色体缺失和获得分析表明,有待发现其他与甲状旁腺肿瘤发生相关的基因。