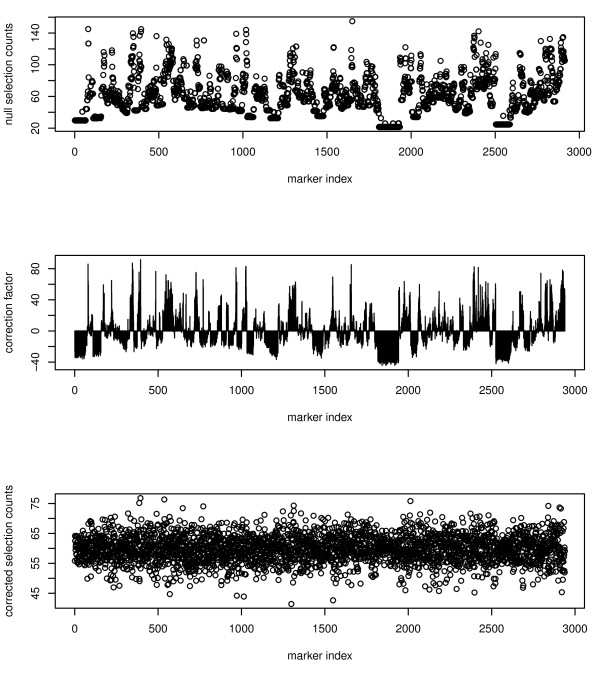
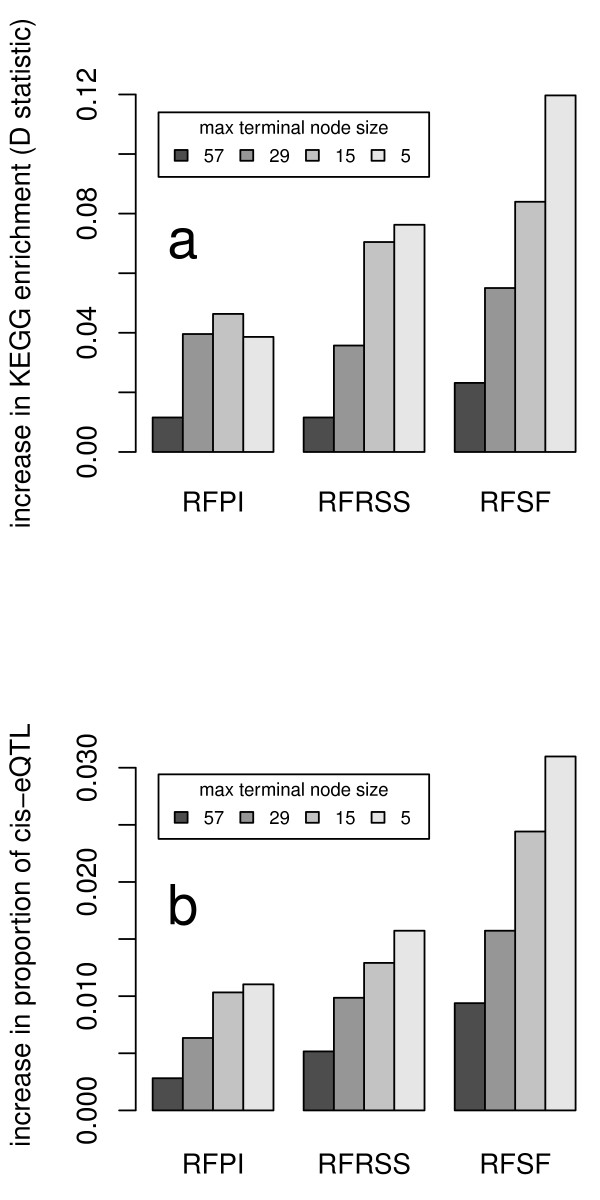
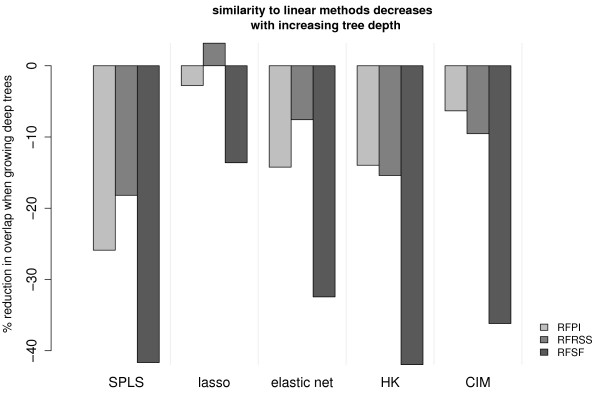
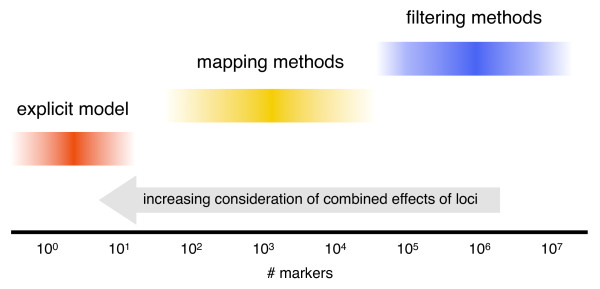
Cellular Networks and Systems Biology, Biotechnology Center - TU Dresden, Dresden, Germany.
BMC Genomics. 2010 Sep 17;11:502. doi: 10.1186/1471-2164-11-502.
The analysis of expression quantitative trait loci (eQTL) is a potentially powerful way to detect transcriptional regulatory relationships at the genomic scale. However, eQTL data sets often go underexploited because legacy QTL methods are used to map the relationship between the expression trait and genotype. Often these methods are inappropriate for complex traits such as gene expression, particularly in the case of epistasis.
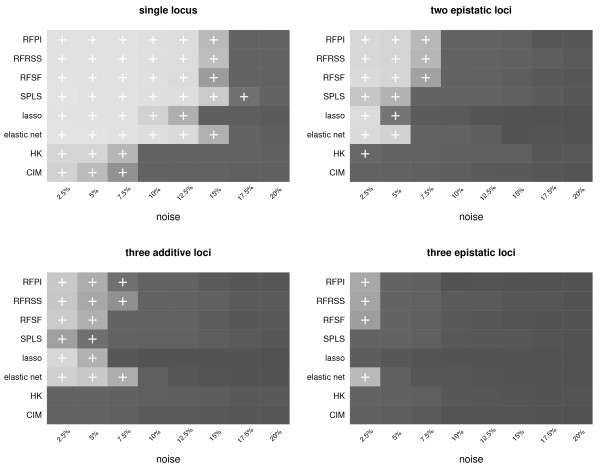
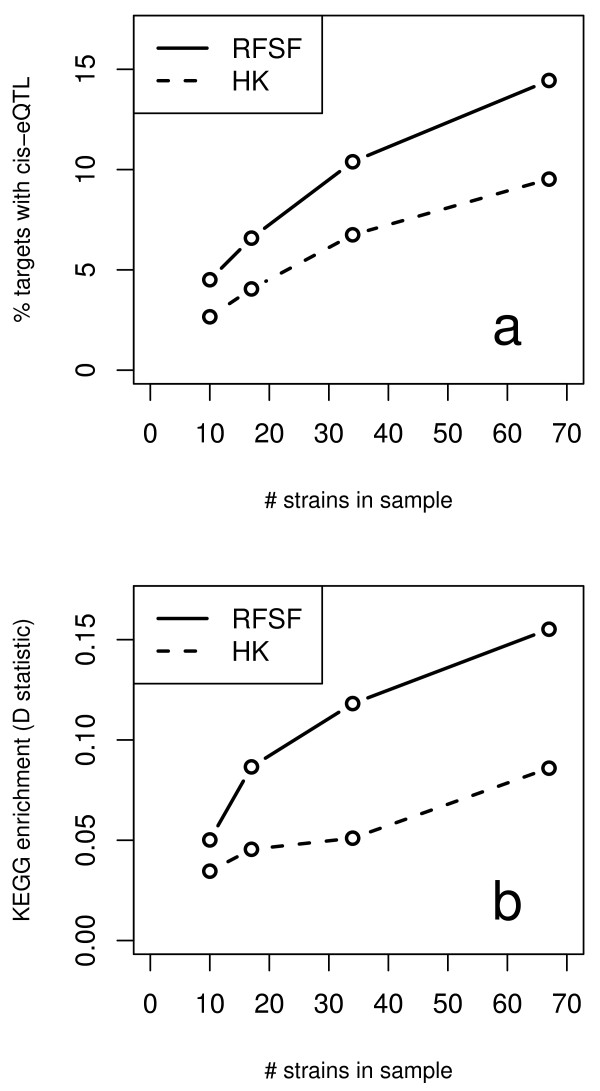
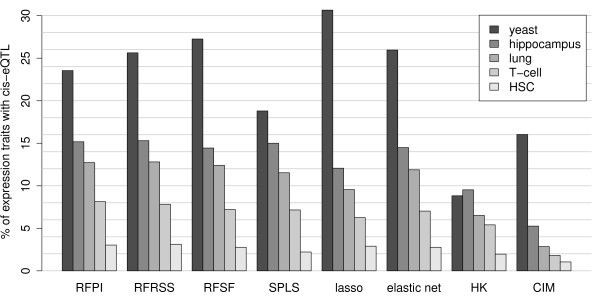
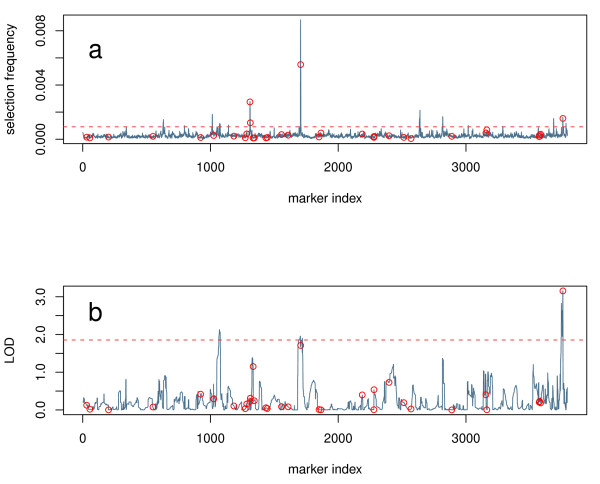
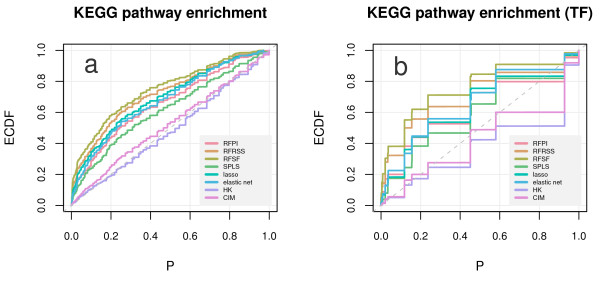
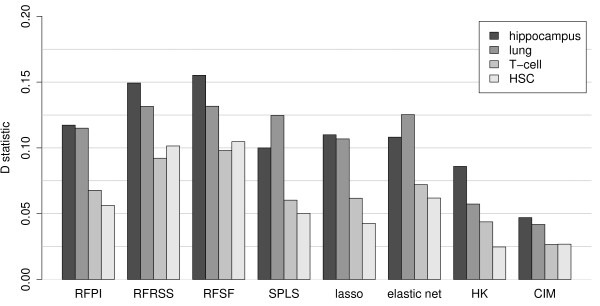
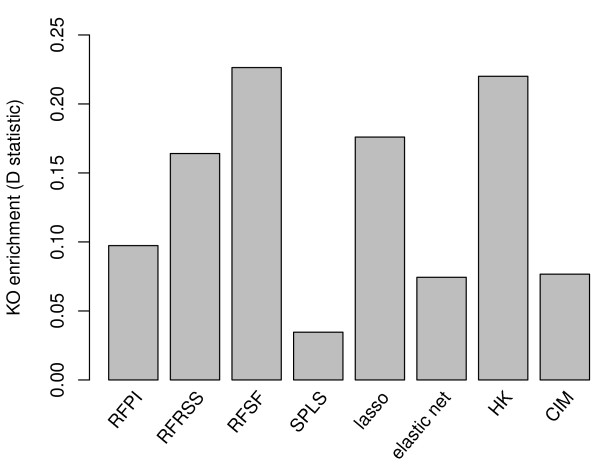
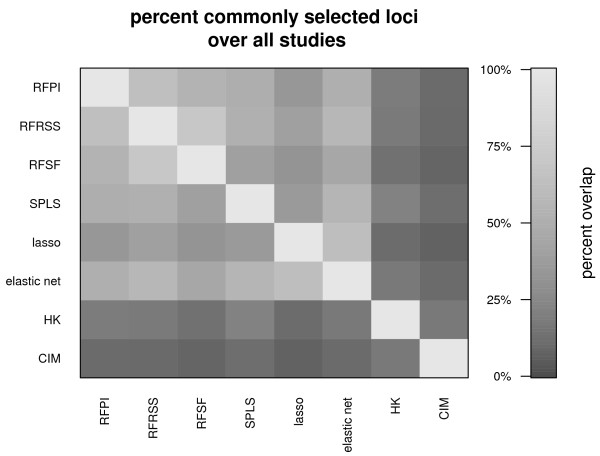
Here we compare legacy QTL mapping methods with several modern multi-locus methods and evaluate their ability to produce eQTL that agree with independent external data in a systematic way. We found that the modern multi-locus methods (Random Forests, sparse partial least squares, lasso, and elastic net) clearly outperformed the legacy QTL methods (Haley-Knott regression and composite interval mapping) in terms of biological relevance of the mapped eQTL. In particular, we found that our new approach, based on Random Forests, showed superior performance among the multi-locus methods.
Benchmarks based on the recapitulation of experimental findings provide valuable insight when selecting the appropriate eQTL mapping method. Our battery of tests suggests that Random Forests map eQTL that are more likely to be validated by independent data, when compared to competing multi-locus and legacy eQTL mapping methods.
表达数量性状基因座(eQTL)分析是在基因组范围内检测转录调控关系的一种潜在强大方法。然而,由于使用传统的 QTL 方法来映射表达性状与基因型之间的关系,eQTL 数据集往往未被充分利用。这些方法通常不适合复杂性状,例如基因表达,特别是在存在上位性的情况下。
在这里,我们将传统的 QTL 映射方法与几种现代多基因座方法进行了比较,并系统地评估了它们在产生与独立外部数据一致的 eQTL 的能力。我们发现,现代多基因座方法(随机森林、稀疏偏最小二乘法、lasso 和弹性网络)在映射的 eQTL 的生物学相关性方面明显优于传统的 QTL 方法(Haley-Knott 回归和复合区间作图)。特别是,我们发现我们基于随机森林的新方法在多基因座方法中表现出优越的性能。
基于重现实验结果的基准为选择适当的 eQTL 映射方法提供了有价值的见解。与竞争的多基因座和传统的 eQTL 映射方法相比,我们的一系列测试表明,随机森林映射的 eQTL 更有可能通过独立数据进行验证。