John P. Hussman Institute for Human Genomics and the Dr. John T. Macdonald Foundation Department of Human Genetics, Miller School of Medicine, University of Miami, Miami, Florida, USA.
Bioinformatics. 2010 Nov 15;26(22):2803-10. doi: 10.1093/bioinformatics/btq526. Epub 2010 Sep 21.
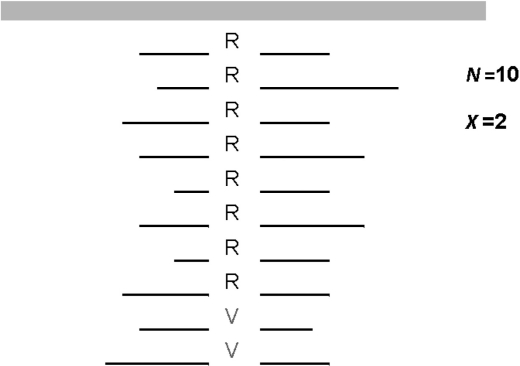
Next-generation sequencing presents several statistical challenges, with one of the most fundamental being determining an individual's genotype from multiple aligned short read sequences at a position. Some simple approaches for genotype calling apply fixed filters, such as calling a heterozygote if more than a specified percentage of the reads have variant nucleotide calls. Other genotype-calling methods, such as MAQ and SOAPsnp, are implementations of Bayes classifiers in that they classify genotypes using posterior genotype probabilities.
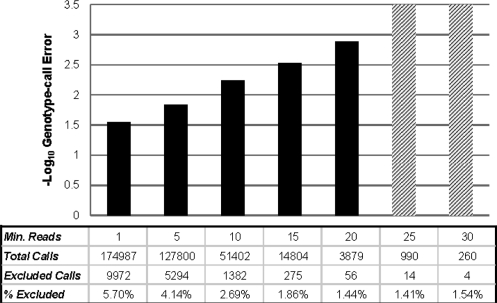
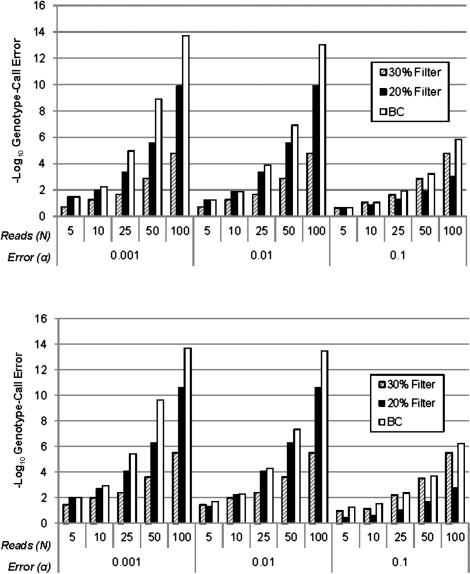
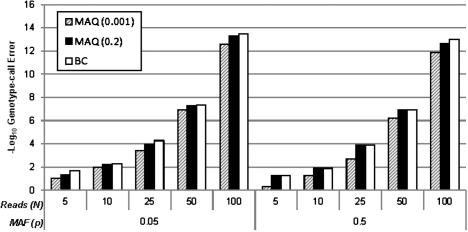
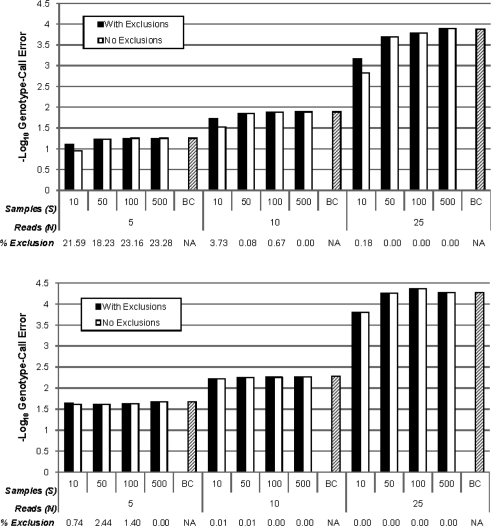
Here, we propose a novel genotype-calling algorithm that, in contrast to the other methods, estimates parameters underlying the posterior probabilities in an adaptive way rather than arbitrarily specifying them a priori. The algorithm, which we call SeqEM, applies the well-known Expectation-Maximization algorithm to an appropriate likelihood for a sample of unrelated individuals with next-generation sequence data, leveraging information from the sample to estimate genotype probabilities and the nucleotide-read error rate. We demonstrate using analytic calculations and simulations that SeqEM results in genotype-call error rates as small as or smaller than filtering approaches and MAQ. We also apply SeqEM to exome sequence data in eight related individuals and compare the results to genotypes from an Illumina SNP array, showing that SeqEM behaves well in real data that deviates from idealized assumptions.
SeqEM offers an improved, robust and flexible genotype-calling approach that can be widely applied in the next-generation sequencing studies.
Software for SeqEM is freely available from our website: www.hihg.org under Software Download.
下一代测序技术提出了几个统计挑战,其中最基本的一个是从一个位置的多个对齐的短读序列中确定个体的基因型。一些简单的基因型调用方法应用固定的过滤器,例如,如果超过指定百分比的读具有变异核苷酸调用,则将杂合子调用。其他基因型调用方法,如 MAQ 和 SOAPsnp,是贝叶斯分类器的实现,因为它们使用后验基因型概率对基因型进行分类。
在这里,我们提出了一种新的基因型调用算法,与其他方法不同,它以自适应的方式估计后验概率的参数,而不是任意地先验指定它们。该算法,我们称之为 SeqEM,将著名的期望最大化算法应用于具有下一代序列数据的无关个体样本的适当似然,利用来自样本的信息来估计基因型概率和核苷酸读取错误率。我们通过分析计算和模拟证明,SeqEM 导致的基因型调用错误率与过滤方法和 MAQ 一样小或更小。我们还将 SeqEM 应用于八个相关个体的外显子序列数据,并将结果与来自 Illumina SNP 数组的基因型进行比较,表明 SeqEM 在偏离理想化假设的真实数据中表现良好。
SeqEM 提供了一种改进的、稳健的和灵活的基因型调用方法,可广泛应用于下一代测序研究中。
SeqEM 的软件可在我们的网站上免费获得:www.hihg.org 下的软件下载。