Leclerc Gilles M, Leclerc Guy J, Fu Guilian, Barredo Julio C
Department of Pediatric Hematology-Oncology, University of Miami Miller School of Medicine, 1601 N,W, 12th Avenue, Miami, FL 33136 USA.
J Mol Signal. 2010 Sep 23;5:15. doi: 10.1186/1750-2187-5-15.
Children with Acute Lymphoblastic Leukemia (ALL) diagnosed with resistant phenotypes and those who relapse have a dismal prognosis for cure. In search for novel treatment strategies, we identified the AMP activated protein kinase (AMPK) as a potential drug target based on its effects on cell growth and survival. We have shown previously that AICAR-induced AMPK activation also induced a compensatory survival mechanism via PI3K/Akt signaling.
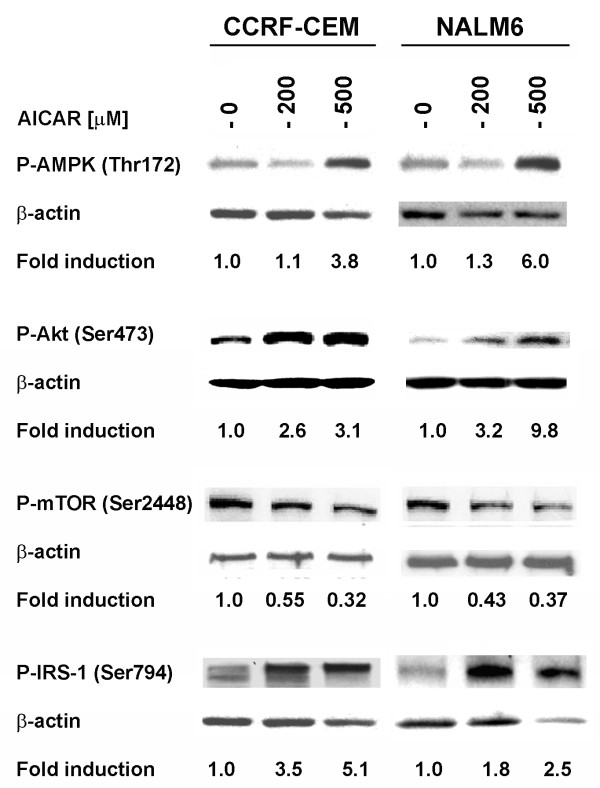
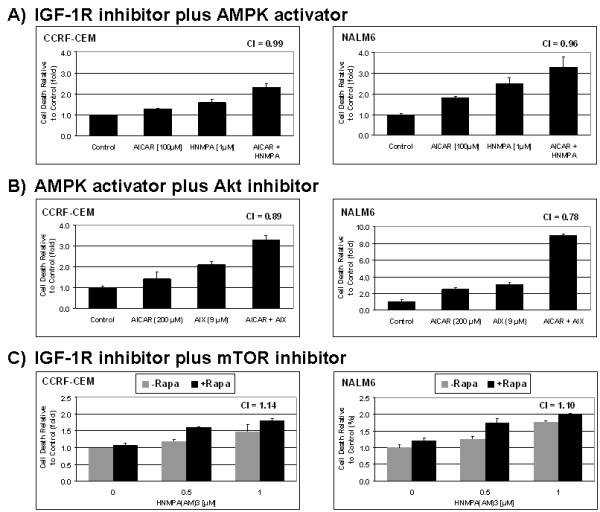
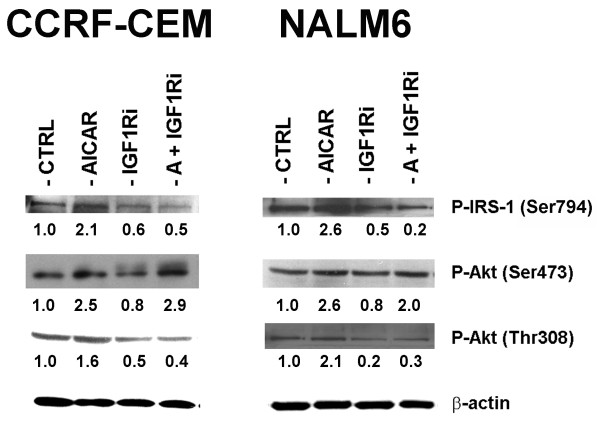
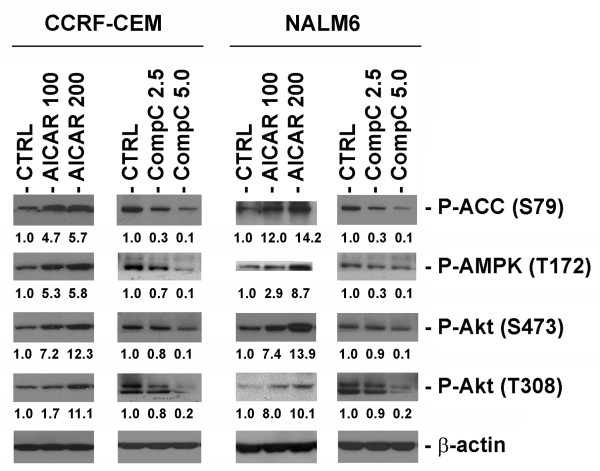
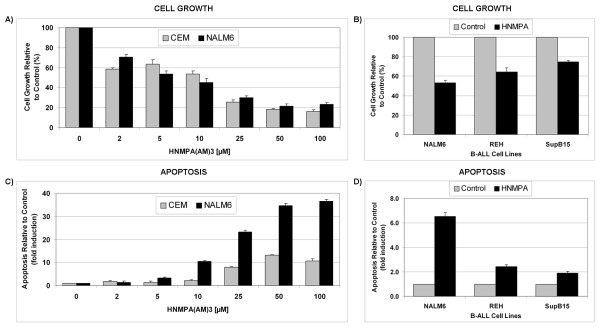
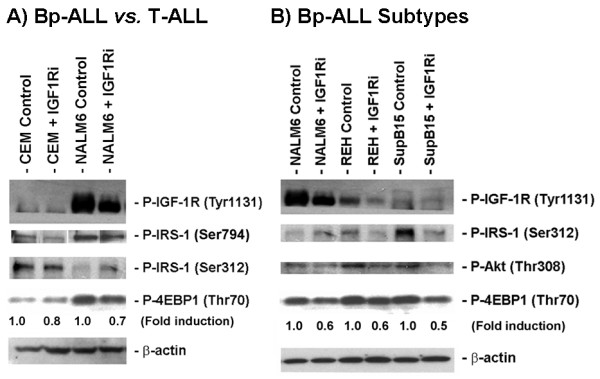
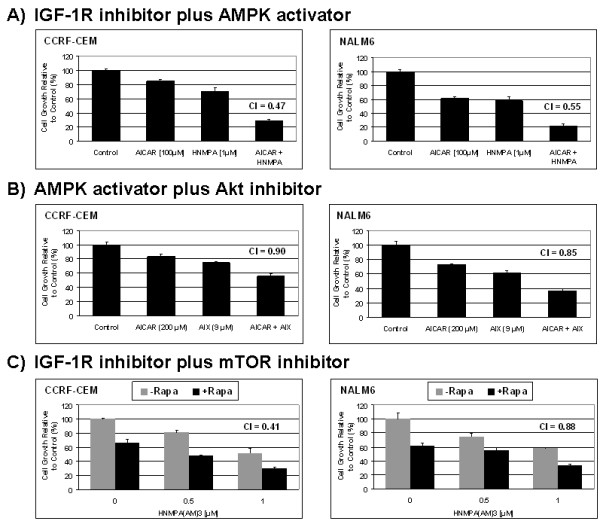
In the present study, we further investigated the downstream signaling induced by AMPK activation in ALL cells. We found that AICAR-induced AMPK activation resulted in up-regulation of P-Akt (Ser473 and Thr308) and decrease of P-mTOR (Ser2448) expression and downstream signaling. We determined that activation of P-Akt (Thr308) was mediated by AMPK-induced IGF-1R activation via phosphorylation of the insulin receptor substrate-1 (IRS-1) at Ser794. Inhibition of IGF-1R signaling using the tyrosine kinase inhibitor HNMPA(AM)3 resulted in significant decrease in P-IRS-1 (Ser794) and P-Akt (Thr308). Co-treatment of AICAR plus HNMPA(AM)3 prevented AMPK-induced up-regulation of P-Akt (Thr308) but did not alter the activation of P-Akt (Ser473). Inhibition of AMPK using compound-C resulted in decreased P-Akt expression at both residues, suggesting a central role for AMPK in Akt activation. In addition, inhibition of IGF-1R signaling in ALL cells resulted in cell growth arrest and apoptosis. Additional Western blots revealed that P-IGF-1R (Tyr1131) and P-IRS-1 (Ser794) levels were higher in NALM6 (Bp-ALL) than CEM (T-ALL), and found differences in IGF-1R signaling within Bp-ALL cell line models NALM6, REH (TEL-AML1, [t(12;21)]), and SupB15 (BCR-ABL, [t(9;22)]). In these models, higher sensitivity to IGF-1R inhibitors correlated with increased levels of IGF-1R expression. Combined therapy simultaneously targeting IGF-1R, AMPK, Akt, and mTOR pathways resulted in synergistic growth inhibition and cell death.
Our study demonstrates that AMPK activates Akt through IGF-1R dependent and independent mechanisms. Co-targeting IGF-1R and related downstream metabolic and oncogenic signaling pathways represent a potential strategy for future translation into novel ALL therapies.
诊断为耐药表型的急性淋巴细胞白血病(ALL)患儿以及复发患儿的治愈预后不佳。为寻找新的治疗策略,基于其对细胞生长和存活的影响,我们确定AMP激活的蛋白激酶(AMPK)为潜在的药物靶点。我们之前已经表明,AICAR诱导的AMPK激活还通过PI3K/Akt信号传导诱导了一种代偿性存活机制。
在本研究中,我们进一步研究了ALL细胞中AMPK激活诱导的下游信号传导。我们发现,AICAR诱导的AMPK激活导致P-Akt(Ser473和Thr308)上调以及P-mTOR(Ser2448)表达和下游信号传导降低。我们确定P-Akt(Thr308)的激活是由AMPK诱导的IGF-1R激活介导的,该激活通过胰岛素受体底物-1(IRS-1)在Ser794处的磷酸化实现。使用酪氨酸激酶抑制剂HNMPA(AM)3抑制IGF-1R信号传导导致P-IRS-1(Ser794)和P-Akt(Thr308)显著降低。AICAR与HNMPA(AM)3联合处理可防止AMPK诱导的P-Akt(Thr308)上调,但不会改变P-Akt(Ser473)的激活。使用化合物C抑制AMPK导致两个位点的P-Akt表达降低,表明AMPK在Akt激活中起核心作用。此外,抑制ALL细胞中的IGF-1R信号传导导致细胞生长停滞和凋亡。额外的蛋白质免疫印迹显示,NALM6(Bp-ALL)中的P-IGF-1R(Tyr1131)和P-IRS-1(Ser794)水平高于CEM(T-ALL),并且在Bp-ALL细胞系模型NALM6、REH(TEL-AML1,[t(12;21)])和SupB15(BCR-ABL,[t(9;22)])中发现了IGF-1R信号传导的差异。在这些模型中,对IGF-1R抑制剂的更高敏感性与IGF-1R表达水平的增加相关。同时靶向IGF-1R、AMPK、Akt和mTOR途径的联合治疗导致协同生长抑制和细胞死亡。
我们的研究表明,AMPK通过依赖IGF-1R和不依赖IGF-1R的机制激活Akt。共同靶向IGF-1R及相关的下游代谢和致癌信号通路代表了一种未来转化为新型ALL治疗方法的潜在策略。