Department of Biology, Centre for Molecular Bioinformatics, University of Rome Tor Vergata, Via della Ricerca Scientifica snc, 00133 Rome, Italy.
Nucleic Acids Res. 2011 Mar;39(4):1231-42. doi: 10.1093/nar/gkq987. Epub 2010 Oct 24.
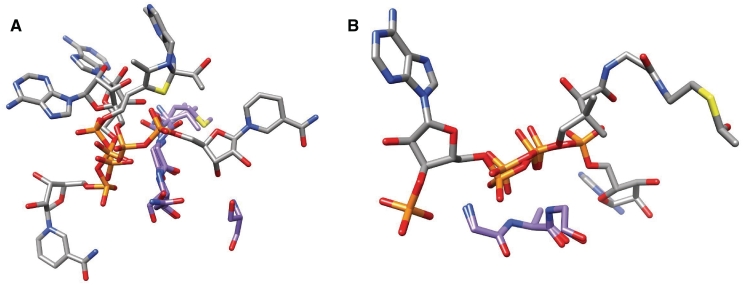
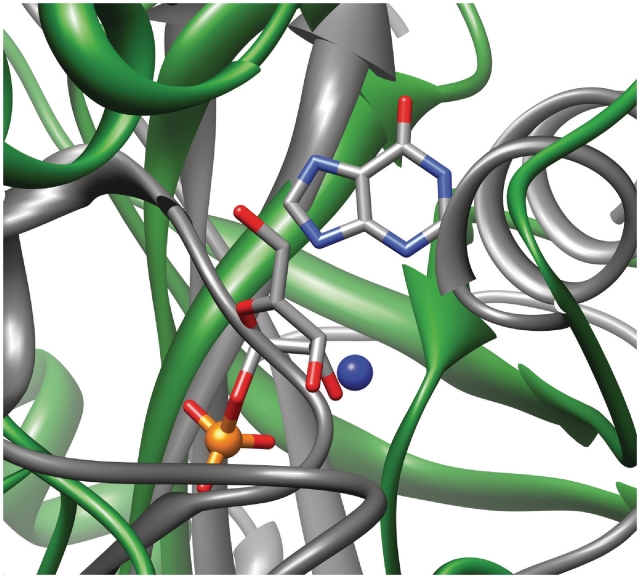
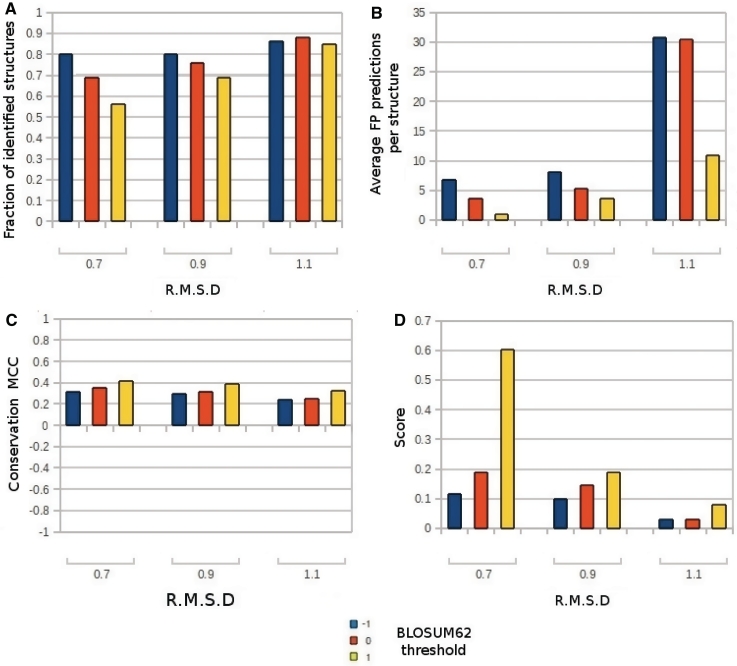
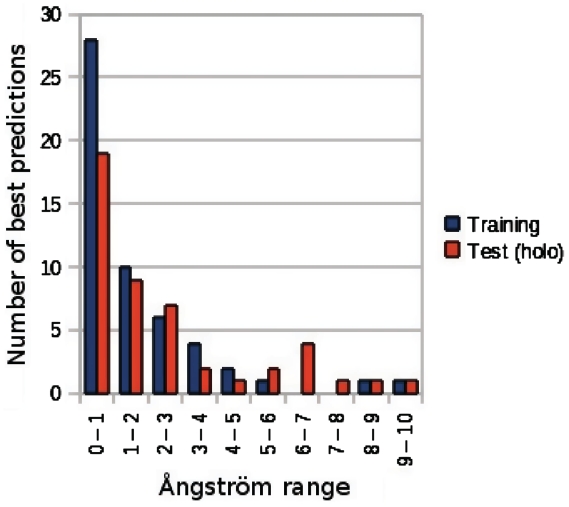
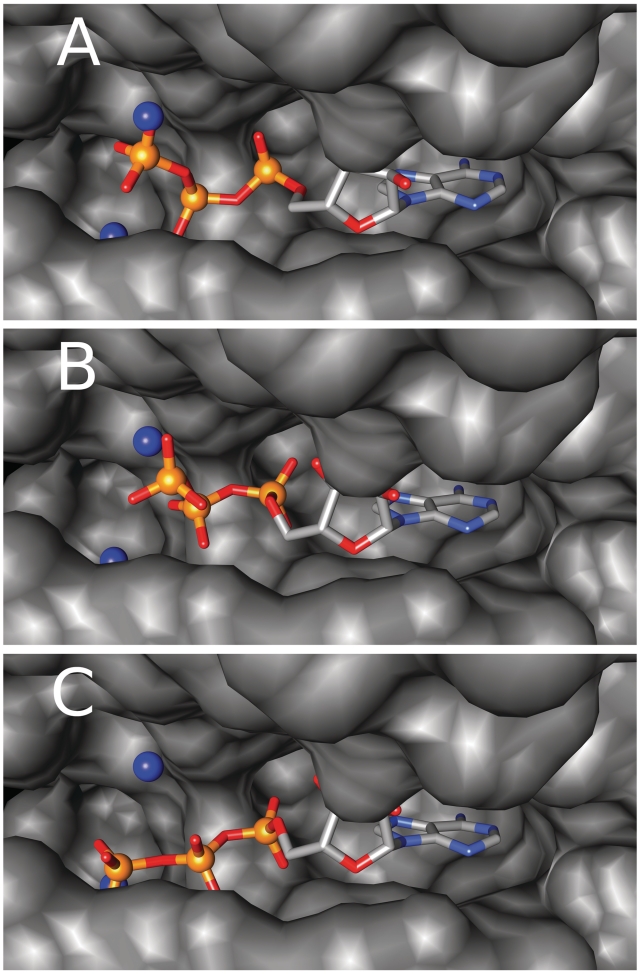
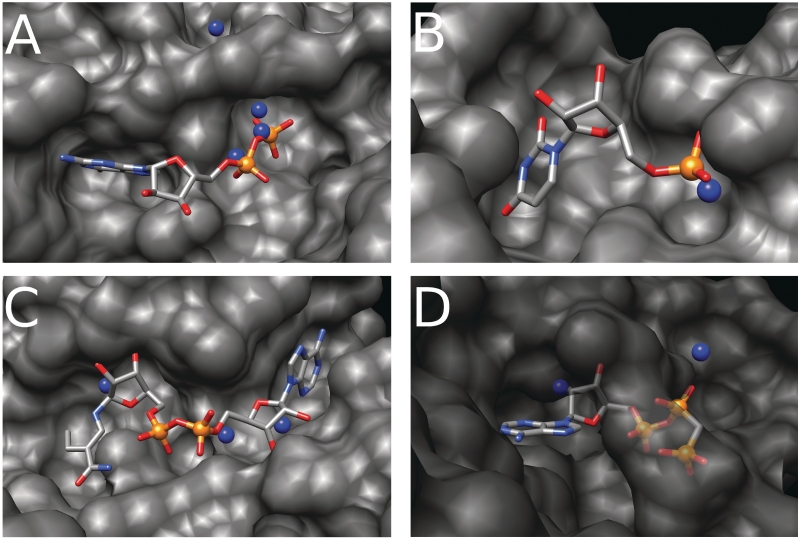
Nearly half of known protein structures interact with phosphate-containing ligands, such as nucleotides and other cofactors. Many methods have been developed for the identification of metal ions-binding sites and some for bigger ligands such as carbohydrates, but none is yet available for the prediction of phosphate-binding sites. Here we describe Pfinder, a method that predicts binding sites for phosphate groups, both in the form of ions or as parts of other non-peptide ligands, in proteins of known structure. Pfinder uses the Query3D local structural comparison algorithm to scan a protein structure for the presence of a number of structural motifs identified for their ability to bind the phosphate chemical group. Pfinder has been tested on a data set of 52 proteins for which both the apo and holo forms were available. We obtained at least one correct prediction in 63% of the holo structures and in 62% of the apo. The ability of Pfinder to recognize a phosphate-binding site in unbound protein structures makes it an ideal tool for functional annotation and for complementing docking and drug design methods. The Pfinder program is available at http://pdbfun.uniroma2.it/pfinder.
已知的蛋白质结构中,近一半的蛋白质结构与含磷酸基团的配体相互作用,如核苷酸和其他辅助因子。已经开发出许多用于识别金属离子结合位点的方法,还有一些用于较大配体(如碳水化合物)的方法,但目前还没有用于预测磷酸结合位点的方法。本文介绍 Pfinder,这是一种预测已知结构蛋白质中磷酸基团结合位点的方法,既可以预测离子形式的磷酸基团结合位点,也可以预测作为其他非肽配体一部分的磷酸基团结合位点。Pfinder 使用 Query3D 局部结构比较算法扫描蛋白质结构,以确定存在一些结构基序,这些基序具有结合磷酸化学基团的能力。我们在一个包含 52 个蛋白质的数据集上测试了 Pfinder,这些蛋白质的apo 和 holo 形式都可用。我们在 63%的 holo 结构和 62%的 apo 结构中至少获得了一个正确的预测。Pfinder 能够识别未结合蛋白质结构中的磷酸结合位点,这使其成为功能注释以及补充对接和药物设计方法的理想工具。Pfinder 程序可在 http://pdbfun.uniroma2.it/pfinder 获得。