Department of Computer Science, Rutgers University, Piscataway, NJ 08854, USA.
BMC Bioinformatics. 2010 Oct 26;11 Suppl 8(Suppl 8):S1. doi: 10.1186/1471-2105-11-S8-S1.
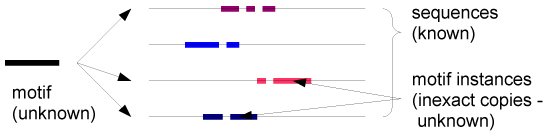
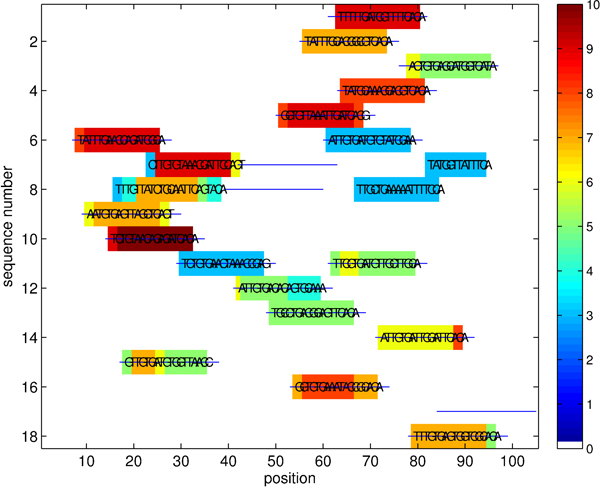
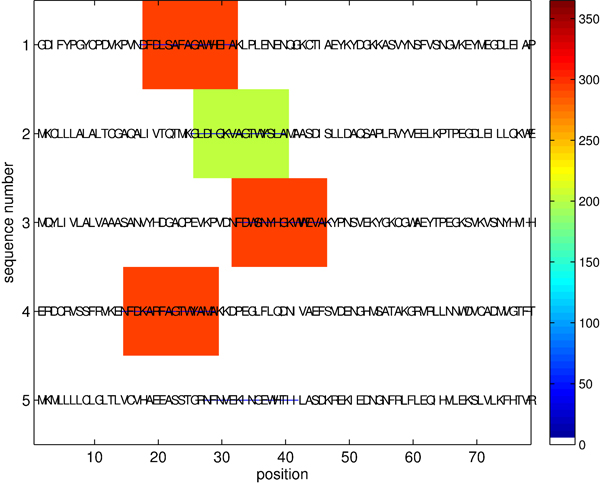
We consider the problem of identifying motifs, recurring or conserved patterns, in the biological sequence data sets. To solve this task, we present a new deterministic algorithm for finding patterns that are embedded as exact or inexact instances in all or most of the input strings.
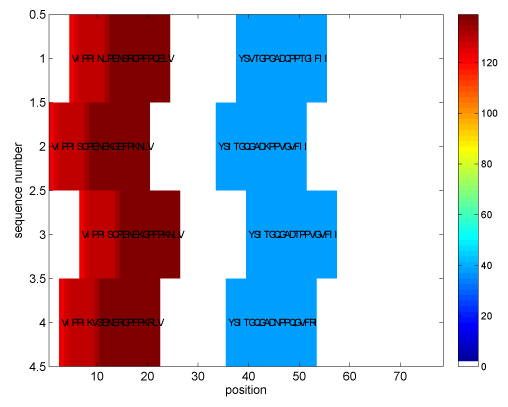
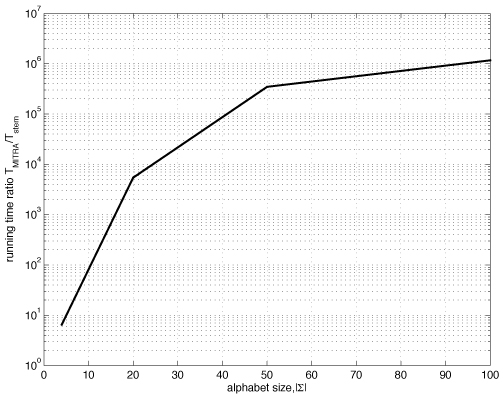
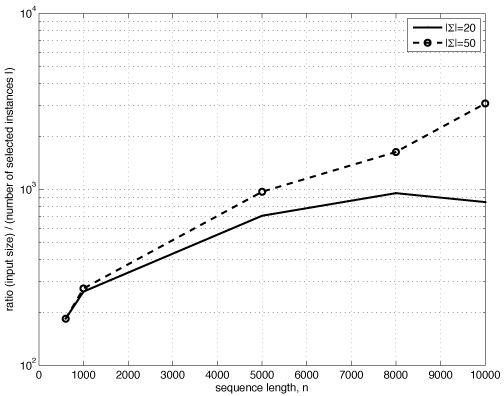
The proposed algorithm (1) improves search efficiency compared to existing algorithms, and (2) scales well with the size of alphabet. On a synthetic planted DNA motif finding problem our algorithm is over 10× more efficient than MITRA, PMSPrune, and RISOTTO for long motifs. Improvements are orders of magnitude higher in the same setting with large alphabets. On benchmark TF-binding site problems (FNP, CRP, LexA) we observed reduction in running time of over 12×, with high detection accuracy. The algorithm was also successful in rapidly identifying protein motifs in Lipocalin, Zinc metallopeptidase, and supersecondary structure motifs for Cadherin and Immunoglobin families.
Our algorithm reduces computational complexity of the current motif finding algorithms and demonstrate strong running time improvements over existing exact algorithms, especially in important and difficult cases of large-alphabet sequences.
我们考虑在生物序列数据集识别基序(重复或保守模式)的问题。为了解决这个任务,我们提出了一种新的确定性算法,用于寻找作为精确或不精确实例嵌入在所有或大多数输入字符串中的模式。
与现有算法相比,所提出的算法(1)提高了搜索效率,(2)与字母表的大小成正比。在合成种植 DNA 基序发现问题上,我们的算法对于长基序比 MITRA、PMSPrune 和 RISOTTO 效率高 10 倍以上。在相同的设置下,字母表较大时,改进幅度更高。在基准 TF 结合位点问题(FNP、CRP、LexA)上,我们观察到运行时间减少了 12 倍以上,并且具有很高的检测准确性。该算法还成功地快速识别了 Lipocalin、Zinc metallopeptidase 以及 Cadherin 和 Immunoglobin 家族的超二级结构基序中的蛋白质基序。
我们的算法降低了当前基序发现算法的计算复杂度,并展示了与现有精确算法相比的强运行时间改进,特别是在大型字母序列的重要和困难情况下。