Zhang Ningzhe, An Mahru C, Montoro Daniel, Ellerby Lisa M
Buck Institute for Age Research and Stanford University Medical School.
PLoS Curr. 2010 Oct 28;2:RRN1193. doi: 10.1371/currents.RRN1193.
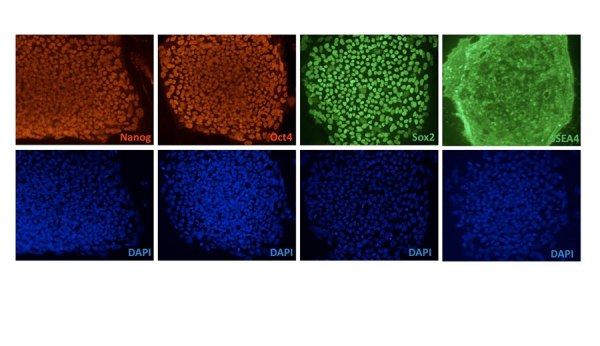
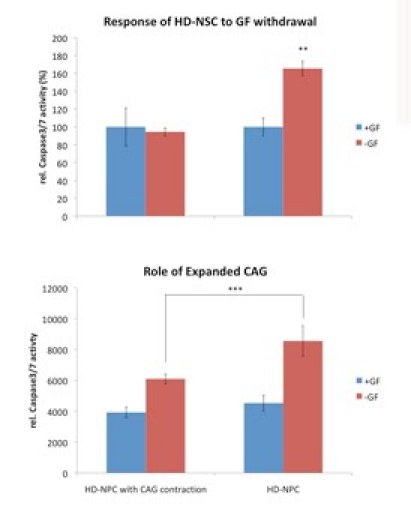
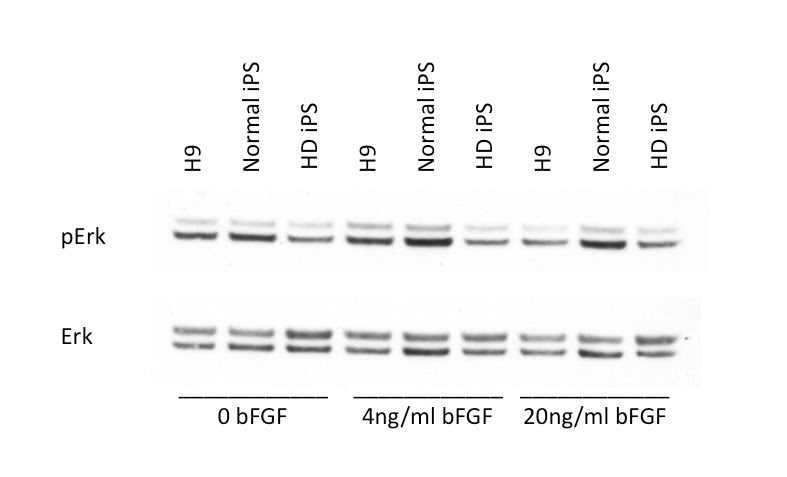
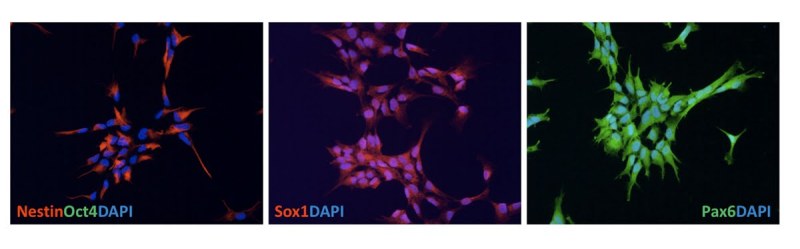
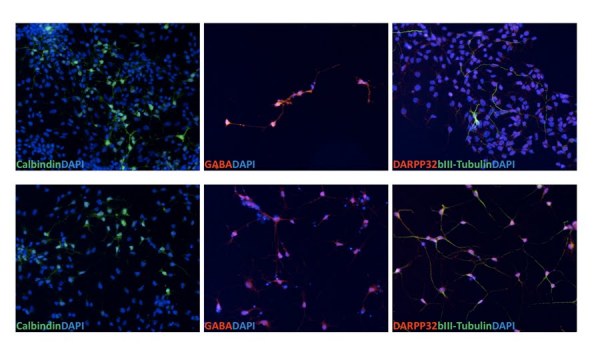
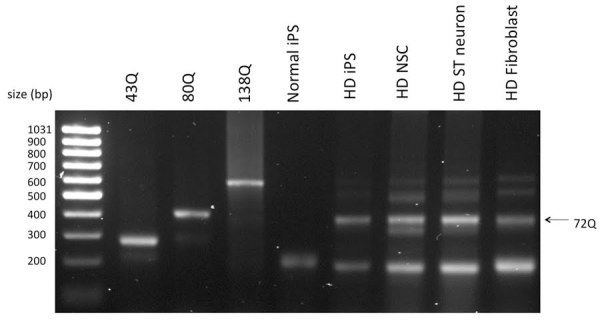
Huntington's disease (HD) is a dominantly inherited neurodegenerative disease caused by a CAG repeat expansion in the first exon of the gene Huntingtin (Htt). A dramatic pathological change in HD is the massive loss of striatal neurons as the disease progresses. A useful advance in HD would be the generation of a human-derived HD model to use for drug screening and understanding mechanisms of HD. We utilized the recently established human iPS cell line derived from HD patient fibroblasts to derive neuronal precursors and human striatal neurons. To achieve this goal, the differentiation of the HD-iPS cells into striatal fate required several steps. First, we generated nestin+/PAX6+/SOX1+/OCT4- neural stem cells (NSCs) from HD-iPS cells using the method of embryoid body formation. HD-NSCs were then subjected to a differentiation condition combining morphogens and neurotrophins to induce striatal lineage commitment. Striatal neuronal precursors/immature neurons stained with β-III tubulin, calbindin and GABA but not DARPP-32 (dopamine- and cyclic AMP-regulated phosphoprotein, Mr = 32,000) were produced in this step. Finally, maturation and terminal differentiation of the striatal neuronal precursors/immature neurons resulted in striatal neurons expressing markers like DARPP-32. The HD-iPS cells derived striatal neurons and neuronal precursors contain the same CAG expansion as the mutation in the HD patient from whom the iPS cell line was established. Moreover, the HD-NSCs showed enhanced caspase activity upon growth factor deprivation compared to normal NSCs (from iPS or H9 NSCs). Therefore, these differentiated cells may produce a human HD cell model useful in the study of HD mechanisms and drug screening.
亨廷顿舞蹈症(HD)是一种常染色体显性遗传的神经退行性疾病,由亨廷顿蛋白(Htt)基因第一外显子中的CAG重复序列扩增引起。随着疾病进展,HD的一个显著病理变化是纹状体神经元大量丧失。HD研究中的一个有益进展是建立用于药物筛选和理解HD发病机制的人源HD模型。我们利用最近建立的源自HD患者成纤维细胞的人诱导多能干细胞系,诱导分化得到神经元前体细胞和人纹状体神经元。为实现这一目标,将HD诱导多能干细胞分化为纹状体细胞需要多个步骤。首先,我们采用胚状体形成法从HD诱导多能干细胞中生成巢蛋白阳性/配对盒蛋白6阳性/性染色体相关基因1阳性/八聚体结合转录因子4阴性的神经干细胞(NSCs)。然后,将HD神经干细胞置于形态发生素和神经营养因子联合的分化条件下,以诱导其向纹状体谱系定向分化。在此步骤中产生了用β-III微管蛋白、钙结合蛋白和γ-氨基丁酸染色,但不被多巴胺和环磷酸腺苷调节的磷蛋白(分子量32,000)染色的纹状体神经元前体细胞/未成熟神经元。最后,纹状体神经元前体细胞/未成熟神经元的成熟和终末分化产生了表达如多巴胺和环磷酸腺苷调节的磷蛋白等标志物的纹状体神经元。源自HD诱导多能干细胞的纹状体神经元和神经元前体细胞含有与建立该诱导多能干细胞系的HD患者突变相同的CAG扩增。此外,与正常神经干细胞(来自诱导多能干细胞或H9神经干细胞)相比,HD神经干细胞在生长因子剥夺后显示出增强的半胱天冬酶活性。因此,这些分化细胞可能会产生一个有助于HD发病机制研究和药物筛选的人源HD细胞模型。