Institute of Animal Husbandry and Breeding, University of Hohenheim, D-70599 Stuttgart, Germany.
Genet Sel Evol. 2010 Nov 1;42(1):40. doi: 10.1186/1297-9686-42-40.
Numerous QTL mapping resource populations are available in livestock species. Usually they are analysed separately, although the same founder breeds are often used. The aim of the present study was to show the strength of analysing F2-crosses jointly in pig breeding when the founder breeds of several F2-crosses are the same.
Three porcine F2-crosses were generated from three founder breeds (i.e. Meishan, Pietrain and wild boar). The crosses were analysed jointly, using a flexible genetic model that estimated an additive QTL effect for each founder breed allele and a dominant QTL effect for each combination of alleles derived from different founder breeds. The following traits were analysed: daily gain, back fat and carcass weight. Substantial phenotypic variation was observed within and between crosses. Multiple QTL, multiple QTL alleles and imprinting effects were considered. The results were compared to those obtained when each cross was analysed separately.
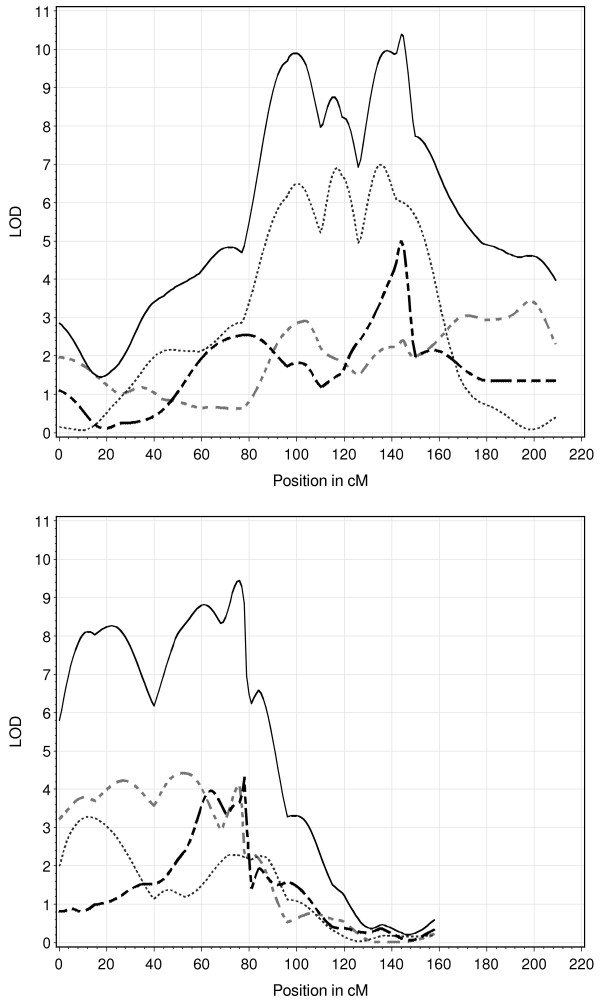
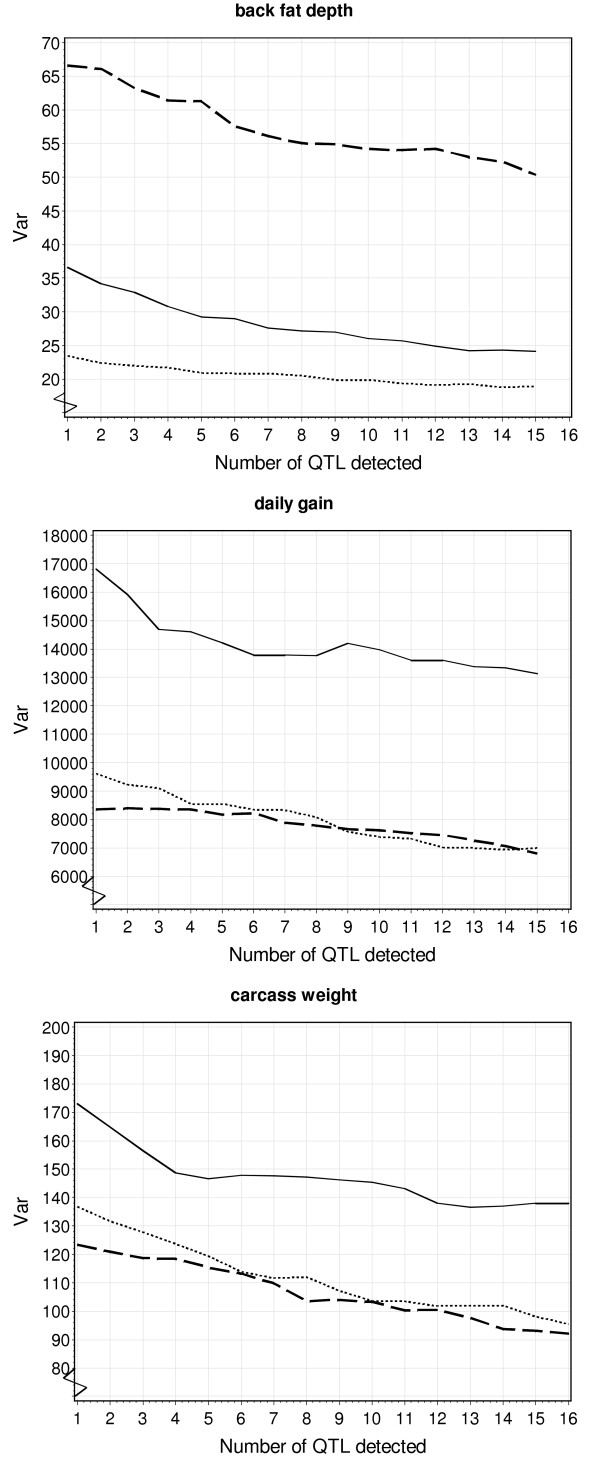
For daily gain, back fat and carcass weight, 13, 15 and 16 QTL were found, respectively. For back fat, daily gain and carcass weight, respectively three, four, and five loci showed significant imprinting effects. The number of QTL mapped was much higher than when each design was analysed individually. Additionally, the test statistic plot along the chromosomes was much sharper leading to smaller QTL confidence intervals. In many cases, three QTL alleles were observed.
The present study showed the strength of analysing three connected F2-crosses jointly. In this experiment, statistical power was high because of the reduced number of estimated parameters and the large number of individuals. The applied model was flexible and was computationally fast.
在畜牧物种中,有许多 QTL 作图资源群体可用。虽然通常会分别分析这些群体,但它们通常使用相同的创始品种。本研究的目的是展示在猪育种中分析几个 F2 杂交群体时的优势,因为几个 F2 杂交群体的创始品种是相同的。
从三个创始品种(即梅山猪、皮特兰猪和野猪)中生成了三个猪 F2 杂交群体。使用灵活的遗传模型联合分析这些杂交群体,该模型估计每个创始品种等位基因的加性 QTL 效应和来自不同创始品种的等位基因组合的显性 QTL 效应。分析了以下性状:日增重、背膘和胴体重量。在杂交群体内部和之间观察到大量表型变异。考虑了多个 QTL、多个 QTL 等位基因和印迹效应。将结果与分别分析每个杂交群体时获得的结果进行了比较。
对于日增重、背膘和胴体重量,分别发现了 13、15 和 16 个 QTL。对于背膘、日增重和胴体重量,分别有三个、四个和五个位点表现出显著的印迹效应。映射的 QTL 数量远高于分别分析每个设计时的数量。此外,染色体上的测试统计图表更加尖锐,导致 QTL 置信区间更小。在许多情况下,观察到了三个 QTL 等位基因。
本研究展示了联合分析三个相关 F2 杂交群体的优势。在本实验中,由于估计参数的数量减少和个体数量增加,统计功效很高。应用的模型灵活且计算速度快。