Thoracic Oncology Section, Surgery Branch, Center for Cancer Research, National Cancer Institute, Bethesda, Maryland, United States of America.
PLoS One. 2010 Oct 29;5(10):e13764. doi: 10.1371/journal.pone.0013764.
Limited information is available regarding mechanisms by which miRNAs contribute to pulmonary carcinogenesis. The present study was undertaken to examine expression and function of miRNAs induced by cigarette smoke condensate (CSC) in normal human respiratory epithelia and lung cancer cells.
Micro-array and quantitative RT-PCR (qRT-PCR) techniques were used to assess miRNA and host gene expression in cultured cells, and surgical specimens. Software-guided analysis, RNA cross-link immunoprecipitation (CLIP), 3' UTR luciferase reporter assays, qRT-PCR, focused super-arrays and western blot techniques were used to identify and confirm targets of miR-31. Chromatin immunoprecipitation (ChIP) techniques were used to evaluate histone marks and transcription factors within the LOC554202 promoter. Cell count and xenograft experiments were used to assess effects of miR-31 on proliferation and tumorigenicity of lung cancer cells.
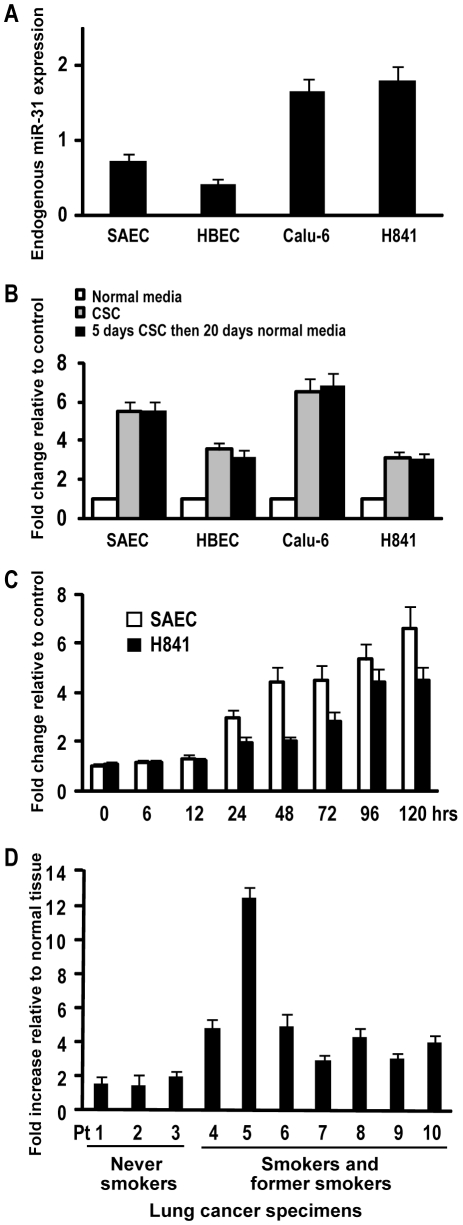
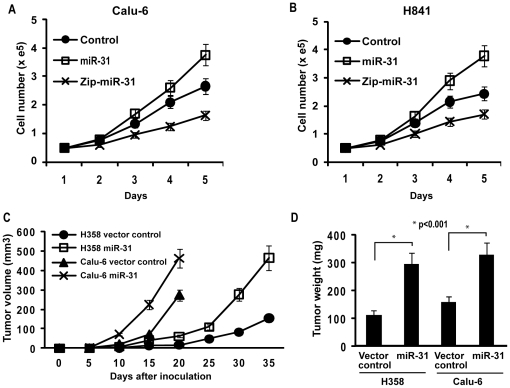
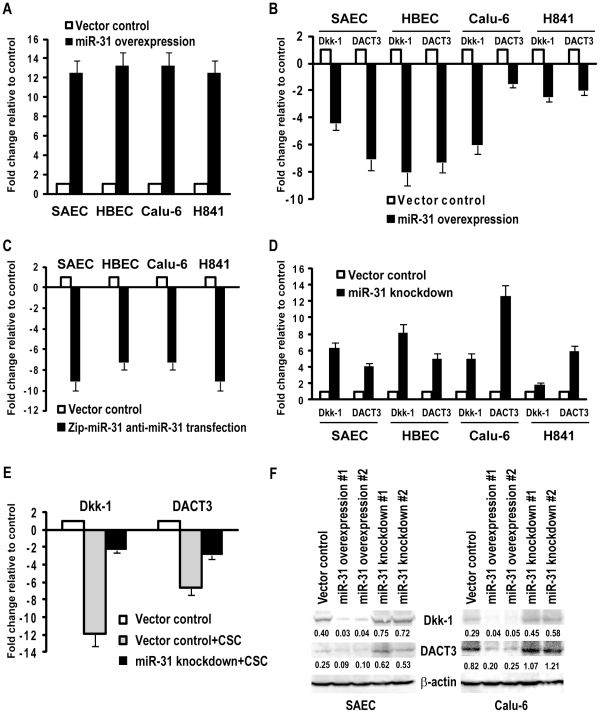
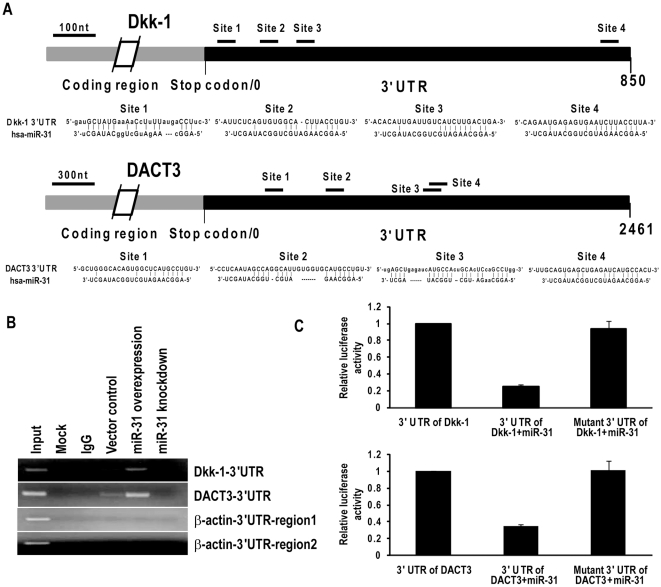
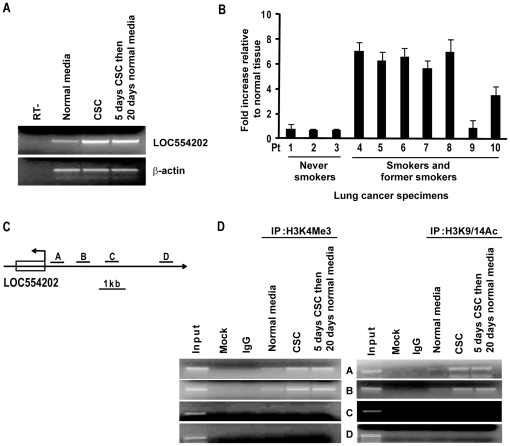
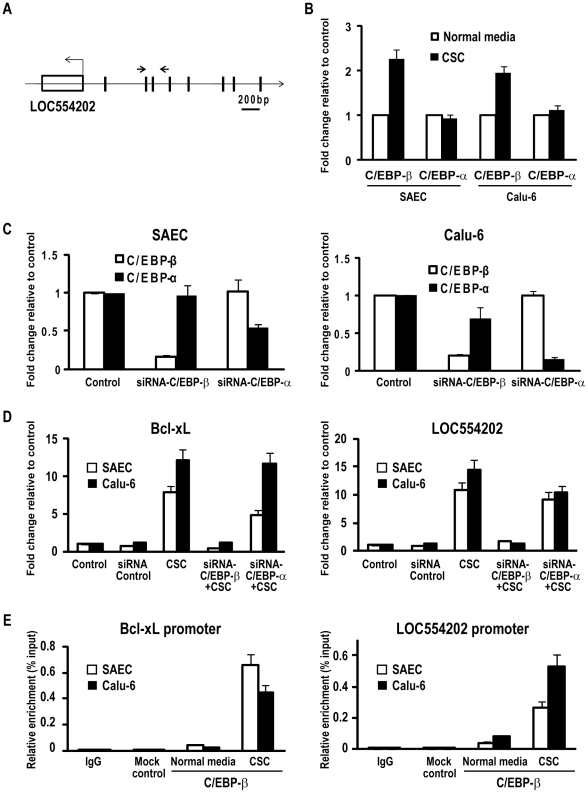
CSC significantly increased miR-31 expression and activated LOC554202 in normal respiratory epithelia and lung cancer cells; miR-31 and LOC554202 expression persisted following discontinuation of CSC exposure. miR-31 and LOC554202 expression levels were significantly elevated in lung cancer specimens relative to adjacent normal lung tissues. CLIP and reporter assays demonstrated direct interaction of miR-31 with Dickkopf-1 (Dkk-1) and DACT-3. Over-expression of miR-31 markedly diminished Dkk-1 and DACT3 expression levels in normal respiratory epithelia and lung cancer cells. Knock-down of miR-31 increased Dkk-1 and DACT3 levels, and abrogated CSC-mediated decreases in Dkk-1 and DACT-3 expression. Furthermore, over-expression of miR-31 diminished SFRP1, SFRP4, and WIF-1, and increased Wnt-5a expression. CSC increased H3K4Me3, H3K9/14Ac and C/EBP-β levels within the LOC554202 promoter. Knock-down of C/EBP-β abrogated CSC-mediated activation of LOC554202. Over-expression of miR-31 significantly enhanced proliferation and tumorigenicity of lung cancer cells; knock-down of miR-31 inhibited growth of these cells.
Cigarette smoke induces expression of miR-31 targeting several antagonists of cancer stem cell signaling in normal respiratory epithelia and lung cancer cells. miR-31 functions as an oncomir during human pulmonary carcinogenesis.
关于 miRNA 如何促进肺癌发生的机制,目前相关信息有限。本研究旨在探讨香烟烟雾冷凝物(CSC)诱导的 miRNA 在正常人呼吸道上皮细胞和肺癌细胞中的表达和功能。
采用微阵列和实时定量 RT-PCR(qRT-PCR)技术检测培养细胞和手术标本中 miRNA 和宿主基因的表达。采用软件指导分析、RNA 交联免疫沉淀(CLIP)、3'UTR 荧光素酶报告基因检测、qRT-PCR、聚焦超级阵列和 Western blot 技术鉴定和确认 miR-31 的靶标。采用染色质免疫沉淀(ChIP)技术评估 LOC554202 启动子内的组蛋白标记物和转录因子。细胞计数和异种移植实验用于评估 miR-31 对肺癌细胞增殖和致瘤性的影响。
CSC 显著增加了正常人呼吸道上皮细胞和肺癌细胞中 miR-31 的表达并激活了 LOC554202;在停止 CSC 暴露后,miR-31 和 LOC554202 的表达仍然持续。与相邻正常肺组织相比,肺癌标本中 miR-31 和 LOC554202 的表达水平显著升高。CLIP 和报告基因检测表明,miR-31 与 Dickkopf-1(Dkk-1)和 DACT-3 直接相互作用。miR-31 的过表达显著降低了正常人呼吸道上皮细胞和肺癌细胞中 Dkk-1 和 DACT3 的表达水平。miR-31 的敲低增加了 Dkk-1 和 DACT3 的水平,并消除了 CSC 介导的 Dkk-1 和 DACT-3 表达降低。此外,miR-31 的过表达降低了 SFRP1、SFRP4 和 WIF-1 的表达,增加了 Wnt-5a 的表达。CSC 增加了 LOC554202 启动子内的 H3K4Me3、H3K9/14Ac 和 C/EBP-β 水平。敲低 C/EBP-β 消除了 CSC 介导的 LOC554202 激活。miR-31 的过表达显著增强了肺癌细胞的增殖和致瘤性;miR-31 的敲低抑制了这些细胞的生长。
香烟烟雾在正常人呼吸道上皮细胞和肺癌细胞中诱导 miR-31 的表达,靶向几种癌症干细胞信号通路的拮抗剂。miR-31 在人类肺致癌作用中作为致癌 miRNA 发挥作用。