de Ronde Jorma J, Klijn Christiaan, Velds Arno, Holstege Henne, Reinders Marcel Jt, Jonkers Jos, Wessels Lodewyk Fa
Department of Bioinformatics and Statistics, The Netherlands Cancer Institute, Plesmanlaan 121, 1066CX Amsterdam, The Netherlands.
BMC Res Notes. 2010 Nov 11;3:298. doi: 10.1186/1756-0500-3-298.
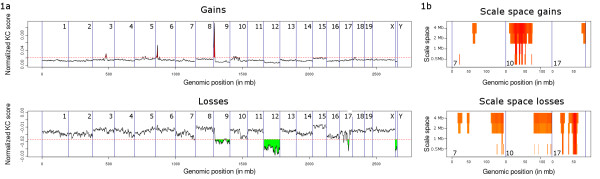
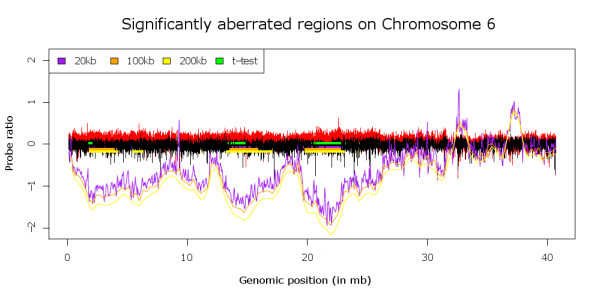
Most approaches used to find recurrent or differential DNA Copy Number Alterations (CNA) in array Comparative Genomic Hybridization (aCGH) data from groups of tumour samples depend on the discretization of the aCGH data to gain, loss or no-change states. This causes loss of valuable biological information in tumour samples, which are frequently heterogeneous. We have previously developed an algorithm, KC-SMART, that bases its estimate of the magnitude of the CNA at a given genomic location on kernel convolution (Klijn et al., 2008). This accounts for the intensity of the probe signal, its local genomic environment and the signal distribution across multiple samples.
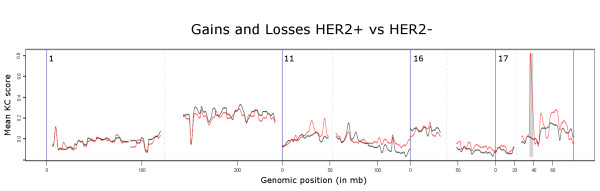
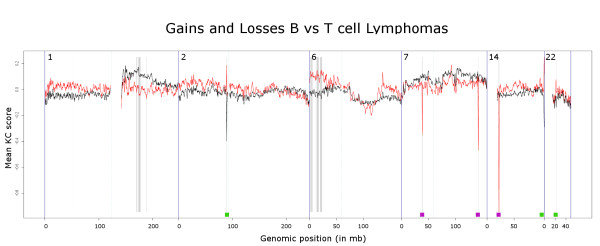
Here we extend the approach to allow comparative analyses of two groups of samples and introduce the R implementation of these two approaches. The comparative module allows for a supervised analysis to be performed, to enable the identification of regions that are differentially aberrated between two user-defined classes.We analyzed data from a series of B- and T-cell lymphomas and were able to retrieve all positive control regions (VDJ regions) in addition to a number of new regions. A t-test employing segmented data, that we implemented, was also able to locate all the positive control regions and a number of new regions but these regions were highly fragmented.
KC-SMARTR offers recurrent CNA and class specific CNA detection, at different genomic scales, in a single package without the need for additional segmentation. It is memory efficient and runs on a wide range of machines. Most importantly, it does not rely on data discretization and therefore maximally exploits the biological information in the aCGH data.The program is freely available from the Bioconductor website http://www.bioconductor.org/ under the terms of the GNU General Public License.
大多数用于在肿瘤样本组的阵列比较基因组杂交(aCGH)数据中寻找复发性或差异性DNA拷贝数改变(CNA)的方法,都依赖于将aCGH数据离散化为获得、缺失或无变化状态。这会导致肿瘤样本中宝贵的生物学信息丢失,而肿瘤样本通常是异质性的。我们之前开发了一种算法KC-SMART,它基于核卷积来估计给定基因组位置处CNA的大小(Klijn等人,2008年)。这考虑了探针信号的强度、其局部基因组环境以及多个样本中的信号分布。
在此,我们扩展了该方法以允许对两组样本进行比较分析,并介绍这两种方法的R实现。比较模块允许进行监督分析,以识别两个用户定义类别之间差异异常的区域。我们分析了一系列B细胞和T细胞淋巴瘤的数据,除了一些新区域外,还能够检索到所有阳性对照区域(VDJ区域)。我们实施的使用分段数据的t检验也能够定位所有阳性对照区域和一些新区域,但这些区域高度碎片化。
KC-SMARTR在一个软件包中提供了不同基因组尺度下的复发性CNA和类别特异性CNA检测,无需额外的分割。它内存效率高,可在多种机器上运行。最重要的是,它不依赖于数据离散化,因此最大程度地利用了aCGH数据中的生物学信息。该程序可根据GNU通用公共许可证条款从Bioconductor网站http://www.bioconductor.org/免费获取。