Division of Biostatistics, German Cancer Research Center, Heidelberg, Germany.
BMC Bioinformatics. 2010 Nov 12;11:556. doi: 10.1186/1471-2105-11-556.
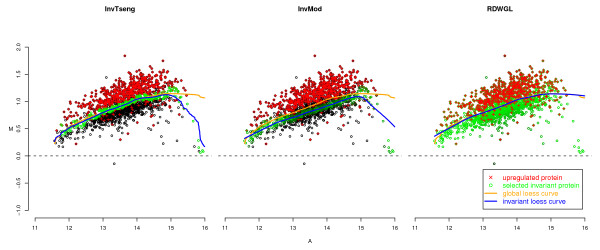
Recent advances in antibody microarray technology have made it possible to measure the expression of hundreds of proteins simultaneously in a competitive dual-colour approach similar to dual-colour gene expression microarrays. Thus, the established normalisation methods for gene expression microarrays, e.g. loess regression, can in principle be applied to protein microarrays. However, the typical assumptions of such normalisation methods might be violated due to a bias in the selection of the proteins to be measured. Due to high costs and limited availability of high quality antibodies, the current arrays usually focus on a high proportion of regulated targets. Housekeeping features could be used to circumvent this problem, but they are typically underrepresented on protein arrays. Therefore, it might be beneficial to select invariant features among the features already represented on available arrays for normalisation by a dedicated selection algorithm.
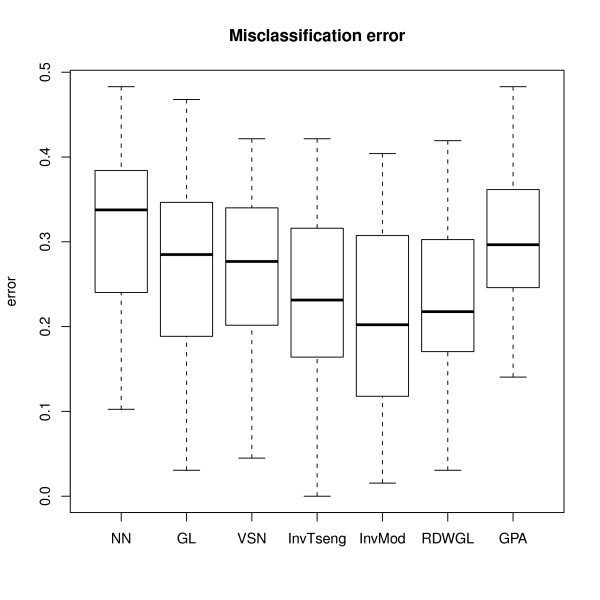
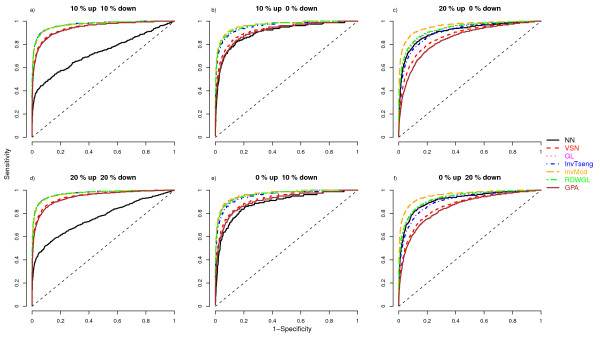
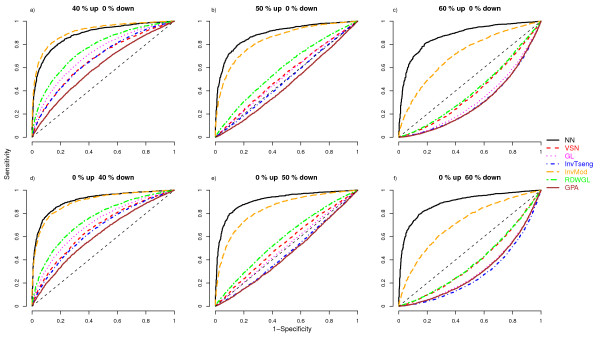
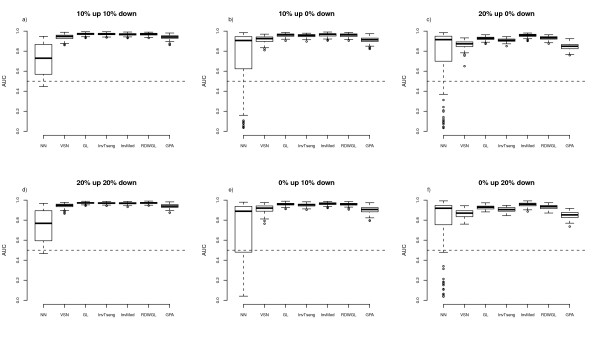
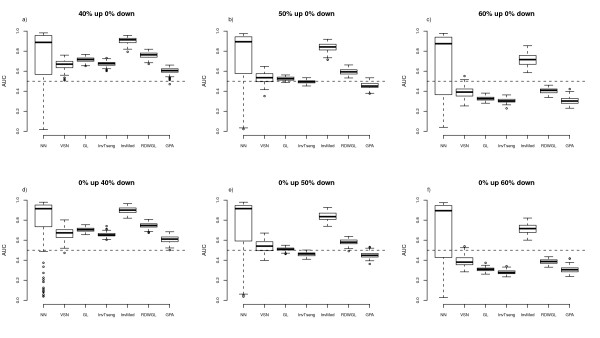
We compare the performance of several normalisation methods that have been established for dual-colour gene expression microarrays. The focus is on an invariant selection algorithm, for which effective improvements are proposed. In a simulation study the performances of the different normalisation methods are compared with respect to their impact on the ability to correctly detect differentially expressed features. Furthermore, we apply the different normalisation methods to a pancreatic cancer data set to assess the impact on the classification power.
The simulation study and the data application demonstrate the superior performance of the improved invariant selection algorithms in comparison to other normalisation methods, especially in situations where the assumptions of the usual global loess normalisation are violated.
抗体微阵列技术的最新进展使得以类似于双色基因表达微阵列的竞争性双色方法同时测量数百种蛋白质的表达成为可能。因此,基因表达微阵列的既定标准化方法,例如局部加权回归,可以原则上应用于蛋白质微阵列。然而,由于在选择要测量的蛋白质时存在偏差,这种标准化方法的典型假设可能会被违反。由于高成本和高质量抗体的有限可用性,当前的阵列通常侧重于高度调控的靶标。管家功能可以用来解决这个问题,但是它们在蛋白质阵列上的代表性通常不足。因此,通过专门的选择算法,选择在现有阵列上已经表示的特征中的不变特征进行归一化可能是有益的。
我们比较了几种已为双色基因表达微阵列建立的标准化方法的性能。重点是一种不变选择算法,并针对该算法提出了有效的改进。在模拟研究中,比较了不同归一化方法在正确检测差异表达特征的能力方面的性能。此外,我们将不同的归一化方法应用于胰腺癌数据集,以评估其对分类能力的影响。
模拟研究和数据应用表明,改进的不变选择算法在与其他归一化方法相比具有优越的性能,尤其是在通常的全局局部加权回归归一化的假设被违反的情况下。