College of Information Engineering, Shanghai Maritime University, Shanghai 201306, China.
Molecules. 2010 Nov 12;15(11):8177-92. doi: 10.3390/molecules15118177.
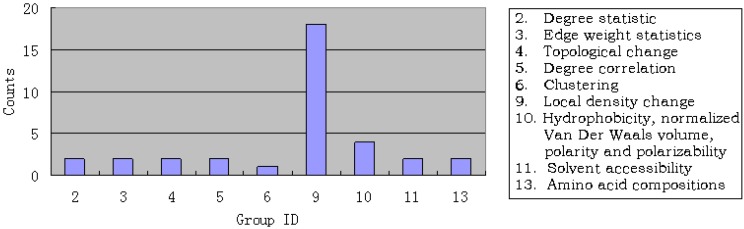
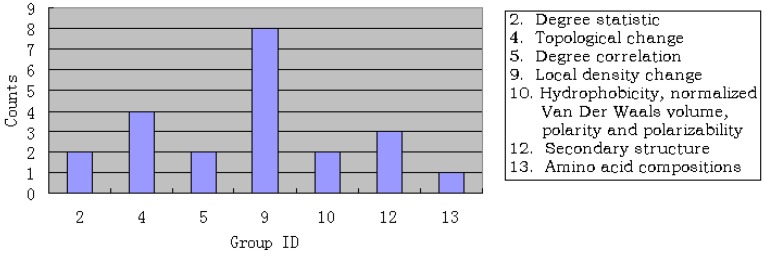
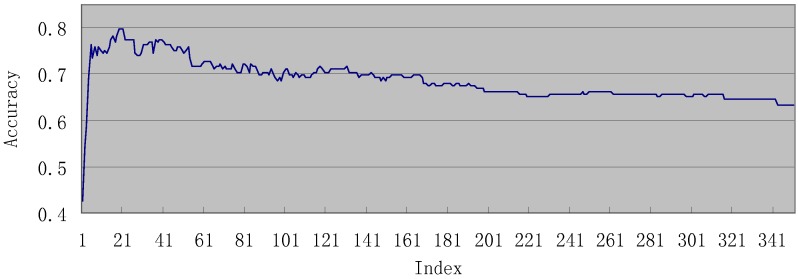
Given a protein-forming system, i.e., a system consisting of certain number of different proteins, can it form a biologically meaningful pathway? This is a fundamental problem in systems biology and proteomics. During the past decade, a vast amount of information on different organisms, at both the genetic and metabolic levels, has been accumulated and systematically stored in various specific databases, such as KEGG, ENZYME, BRENDA, EcoCyc and MetaCyc. These data have made it feasible to address such an essential problem. In this paper, we have analyzed known regulatory pathways in humans by extracting different (biological and graphic) features from each of the 17,069 protein-formed systems, of which 169 are positive pathways, i.e., known regulatory pathways taken from KEGG; while 16,900 were negative, i.e., not formed as a biologically meaningful pathway. Each of these protein-forming systems was represented by 352 features, of which 88 are graph features and 264 biological features. To analyze these features, the "Minimum Redundancy Maximum Relevance" and the "Incremental Feature Selection" techniques were utilized to select a set of 22 optimal features to query whether a protein-forming system is able to form a biologically meaningful pathway or not. It was found through cross-validation that the overall success rate thus obtained in identifying the positive pathways was 79.88%. It is anticipated that, this novel approach and encouraging result, although preliminary yet, may stimulate extensive investigations into this important topic.
给定一个蛋白质形成系统,即由一定数量的不同蛋白质组成的系统,它能否形成有生物学意义的途径?这是系统生物学和蛋白质组学中的一个基本问题。在过去的十年中,大量关于不同生物体的信息,包括遗传和代谢水平的信息,已经被积累并系统地存储在各种特定的数据库中,如 KEGG、ENZYME、BRENDA、EcoCyc 和 MetaCyc。这些数据使得解决这样一个基本问题成为可能。在本文中,我们通过从 17069 个蛋白质形成系统中的每一个系统中提取不同的(生物和图形)特征,来分析人类已知的调节途径,其中 169 个是阳性途径,即从 KEGG 中获取的已知调节途径;而 16900 个是阴性的,即没有形成有生物学意义的途径。每个蛋白质形成系统由 352 个特征表示,其中 88 个是图形特征,264 个是生物特征。为了分析这些特征,我们使用了“最小冗余最大相关性”和“增量特征选择”技术,选择了一组 22 个最佳特征来查询蛋白质形成系统是否能够形成有生物学意义的途径。通过交叉验证发现,由此获得的识别阳性途径的总体成功率为 79.88%。尽管这只是初步的结果,但我们预计这种新的方法和令人鼓舞的结果可能会激发对这个重要主题的广泛研究。